
Abstract
The acute leukemias are the most common cancer of children, adolescents, and young adults. These diseases are characterized by a tremendous variability in clinical course, prompting a continuing search for accurate predictors of outcome. Using algorithms based on clinical features at presentation, response to therapy, and several molecular analyses, some patients are diagnosed with features of high-risk disease and comparatively greater risk for relapse. Molecular analyses of patients with high-risk acute leukemias have resulted in an improved understanding of how dysregulated cellular signaling can affect resistance to conventional therapy. Whereas exciting discoveries continue to be made in the identification of relevant molecular biomarkers and targeted therapies, the challenges and opportunities associated with these findings remain to be clearly defined in future clinical trials.
Introduction: assessing risk for relapse in pediatric acute leukemias
Through improved approaches to risk classification, therapy intensification, and supportive care, children and young adults with pediatric acute leukemias have improved survival rates that greatly exceed those from 50 years ago.1 This gratifying trend has been a testament to the success of risk-based therapies. Despite this progress, relapse and disease progression in patients with high-risk disease remain common problems for which salvage therapies often do not achieve a long-term remission. For children with acute myelogenous leukemia (AML) or acute lymphoblastic leukemia (ALL), a slow response to initial therapy, persistence of measurable disease beyond induction, or relapse after a first remission is associated with a dismal prognosis. In addition to these treatment response–based features of high-risk disease, a growing number of specific, recurring karyotypic features and molecular aberrations may also indicate a high chance to relapse from leukemias that are intrinsically resistant to conventional therapies. In many cases, expression profiling, whole-genome sequencing, and other molecular analyses have provided insight into the signaling pathways that are mechanistically related to chemotherapy resistance. These technologies are being used to search for as-yet-undetected molecular abnormalities and to guide a rationale design of targeted therapies because adequate treatment itself remains the most important prognostic factor of all.
According to data provided by the Surveillance Epidemiology and End Results registry (SEER; http://www.seer.cancer.gov/), AML and ALL are diagnosed in approximately 3600 children and young adults annually in the United States. Similarly, the incidence of childhood leukemia is 46.7 cases/million/year in the World Health Organization European region (www.euro.who.int/ENHIS). Using a combination of response-based, genetically defined, and treatment-era–specific criteria, high-risk disease is identified in approximately 15%-35% of AML2,3 and ALL4–6 patients who are 20 years of age or younger. In the past, these subsets of patients were often assigned comparatively intensified conventional therapy. Whereas this approach has achieved modestly improved event-free survival (EFS) over the past few decades, further intensification has caused additional toxicity without much benefit. Because outcomes for high-risk leukemias appear to have plateaued with conventional therapy, the need for less toxic, “targeted” therapies has become greater, and consequently an increased sophistication in determining which patients might benefit from a targeted approach. The data gained from the Human Genome Project and the technologies used to drive this effort have transformed our understanding of the molecular determinants that define high-risk AML and ALL. In concert with this transformation has been the development of molecularly targeted therapies that are currently achieving exciting improvements in EFS for BCR-ABL–rearranged ALL, setting a new paradigm for other types of high-risk disease. With these successes, important challenges, opportunities, and questions are being raised: Is biological effect the same as maximum tolerated dose? Should a novel agent be used as a single agent or in combination with dose-intensified therapy? Is it feasible to test a targeted therapy in a randomized controlled trial or should it be nonrandomly assigned to those having a particular biomarker? Has the biomarker been reasonably validated for its sensitivity, specificity, and prevalence in a given leukemia cohort?
For pediatric acute leukemias, the molecular determinants of risk have yet to be fully defined, and target therapies remain in the earliest stages of discovery. The importance of clinical trial design and supportive care will need to be revisited in the current treatment era. In the following sections, therapies informed by the molecular analyses of high-risk disease for AML and ALL will be discussed within the context of an evolving research field.
Clinical and molecular features of high-risk disease in pediatric AML
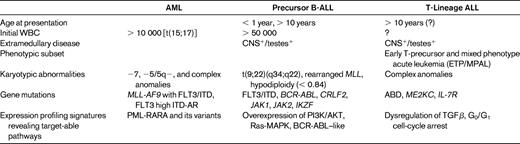
As commonly practiced for ALL, children diagnosed with AML require assignment into risk-stratified treatment groups. Unlike ALL, initial WBC and CNS involvement do not appear to have a similar weight in determining risk status (Table 1); rather, marked differences in cure rates have been observed for various treatment response groups and genetic subsets.3,7 In a report from the United Kingdom's MRC AML 10 clinical trial,3 risk group stratification was further improved with the identification of patient subsets with a complete response (< 5% blasts), partial response (5%-15% blasts), or resistant disease (> 15% blasts) after the first course of induction failure consisting of cytarabine, daunomycin, and etoposide. After further intensification, patients with end-induction complete response, partial response, or resistant disease had overall survival (OS) rates of 53%, 44%, and 22%, respectively.8 Additional studies in pediatric AML have shown that 3 genetically based risk groups have been identified: (1) a favorable risk group consisting of t(15;17)[PML-RARα], t(8;21)[AML1-ETO], and inv16[CBFβ-MYH11]; (2) an intermediate group, which includes MLL chimeric fusion genes (Table 2) or a normal (uninformative) karyotype; and (3) a high-risk group, including −5/del(5q), −7/del(7q), inv3/t,(3,3), +8, and complex karyotypes.8–10 Other high-risk cytogenetic markers include high expression of the RUNX3 gene and low expression of the ATRX gene, but because they have been identified with different technological approaches and within the context of differing treatments, they may be protocol specific.9
Commercially available molecular tests associated with high-risk acute leukemia*

Karyotypic and MRD testing are locally available (either on-site or as a send-out test) at most health care centers; check with your local hematopathologist about the availability of such tests at your institution.
†Disclaimer that test is not intended for the diagnosis, prevention, or treatment of a disease.
Fms-like tyrosine kinase receptor-3 (FLT3) and its ligand are important for normal hematopoiesis. Internal tandem duplications (ITDs) in exons 14 and/or 15 lead to constitutive receptor activation, and “activating loop” mutations involving exon 20 are the most common somatic mutation observed in AML (Table 2). Mutations of FLT affect approximately 20% of children with AML, and are not associated with FAB class or other cytogenetic markers.11 In affected patients, FLT3/ITD mutations are thought to disrupt a repressor sequence in the juxta-membrane region and confer a poor prognosis.11,12 In a retrospective study of children with newly diagnosed AML enrolled in CCG2891, those with FLT3/ITDs had an 8-year OS and EFS rates of 14% and 7%, respectively, compared with 50% and 44% for patients without the FLT3/ITD molecular aberrancy.13 However, a subpopulation of AML patients with FLT3/ITD appear to respond well to conventional therapy,9 leading to the observation that the proportion of the mutant FLT3/ITD to the wild-type varies significantly within a patient population. Patients with an FLT3/ITD allelic ratio (ITD-AR) range from < 0.1 to > 10, and those with high ITD-AR have a significantly higher rate of relapse and are thus considered to be high risk. In blending together these clinical and molecular features, the current high-risk subset of children with AML is composed of those who have > 15% blasts after the first course of induction therapy, those who have monosomy 7, monosomy 5/5q−, or FLT3 high ITD-AR.
Treatment options for high-risk pediatric AML and inhibitors of the FLT3 kinase
After appropriate induction therapy, 75%-90% of all children diagnosed with AML enter into first remission, and approximately 50% are eventually cured of their disease.14 For patients with high-risk disease, remission induction rates are comparatively lower at 70%, and a heightened chance of disease recurrence has been somewhat mitigated by either related or unrelated allogeneic stem cell transplantation in first remission.15 In high-risk patients for whom a suitable donor cannot be found, relapse continues to be a common problem, calling for novel agents or targeted therapies.
The importance of FLT3-kinase signaling in AML has led to the development of agents with selective inhibitory activity. Whereas several FLT3-inhibitory agents are currently under development, CEP-701 (lestaurtinib) has so far received the most extensive studies in AML clinical trials for infants, children, and adults.16 The Children's Oncology Group (COG) recently completed a pilot study of lestaurtinib in combination with cytarabine and idarubicin in relapsed or refractory FLT-3 mutant AML (AAML06P1). This study was designed to investigate the dose-limiting toxicities of lestaurtinib in a nonrandomized setting for patients < 30 years of age. After an initial safety phase, in which lestaurtinib (50 mg/m2 twice daily followed by 62.5 mg/m2 twice daily) was used in combination with cytarabine and idarubicin, the study entered an efficacy phase to assess whether lestaurtinib could be provided in sufficient doses to adequately inhibit FLT3 kinase in patient plasma. This study passed its safety phase evaluation, and recently closed upon completion of its accrual goals for an overall assessment for lestaurtinib efficacy. Currently, 6 other FLT3 inhibitors are under evaluation in clinical trials for AML. These investigational agents include sorafenib, sunitinib, midostaurin (PKC412), lestaurtinib, tandutinib (MLN518), AC220, and KW-2449 (reviewed in Wiernik17 ).
Several investigators have sought to elucidate important signaling pathways in pediatric AML. Cases harboring mutations of the mixed-lineage leukemia gene (MLL; 11q23) with partial tandem duplications and chimeric fusions were identified, but failed to cluster together in expression profiling studies.10 The biphenotypic presentation of MLL-associated leukemias has been associated with stem-cell features and distinct transformation pathways (Table 2 and Figure 1).18 Secondary neoplasms presenting as 11q23 rearranged AML has been a frequently reported complication of cytotoxic therapy, especially associated with epipodophyllotoxin exposure. Interestingly, MLL-rearranged AML may copresent with FLT3 mutations, which has been associated with a particularly aggressive clinical course.19 For more information on this topic, please see the article entitled “Targeted Epigenetic Programs in MLL-Rearranged Leukemias” by Dr. Scott Armstrong.

Karyotypic analysis in a 2-year-old boy who presented with M5a AML and relapsed 6 years later with the same translocation in precursor-B ALL. FISH for the MLL gene demonstrates a positive/rearranged result. This single-interphase nucleus shows the typical abnormal signal pattern for the MLL break-apart FISH probe. The abnormal MLL allele is identified by the distinctly separate (broken apart) orange and green signals (yellow arrow). In contrast, the remaining second MLL allele shows a normal fusion pattern of overlapping green and orange signals. Inset shows a selected G-banded karyotype demonstrating an abnormal karyotype; 46,XY,del(6)(q13),t(9;11)(p22;q23). The translocation involves a reciprocal exchange of genomic material on chromosomes 9 and 11. As a consequence of this translocation, the MLL gene localized to 11q23 is juxtaposed to the MLLT3 gene on 9p22, resulting in an oncogenic chimeric fusion gene. The translocation has been associated with IGSF4 overexpression that is partially regulated by promoter methylation.19 (Images courtesy of Dr. Kaaren Reichard.)
Karyotypic analysis in a 2-year-old boy who presented with M5a AML and relapsed 6 years later with the same translocation in precursor-B ALL. FISH for the MLL gene demonstrates a positive/rearranged result. This single-interphase nucleus shows the typical abnormal signal pattern for the MLL break-apart FISH probe. The abnormal MLL allele is identified by the distinctly separate (broken apart) orange and green signals (yellow arrow). In contrast, the remaining second MLL allele shows a normal fusion pattern of overlapping green and orange signals. Inset shows a selected G-banded karyotype demonstrating an abnormal karyotype; 46,XY,del(6)(q13),t(9;11)(p22;q23). The translocation involves a reciprocal exchange of genomic material on chromosomes 9 and 11. As a consequence of this translocation, the MLL gene localized to 11q23 is juxtaposed to the MLLT3 gene on 9p22, resulting in an oncogenic chimeric fusion gene. The translocation has been associated with IGSF4 overexpression that is partially regulated by promoter methylation.19 (Images courtesy of Dr. Kaaren Reichard.)
Clinical and molecular features of high-risk disease in pediatric ALL
As observed in patients with AML, pediatric ALL is quite varied and heterogeneous in its presentation, biology, and treatment response. Over the past several decades, several clinical features have been identified that help to define a high-risk subset of patients: age < 1 year or > 10 years, initial WBC > 50 000 cells/μL, and the presence of leukemic blasts in the blood-brain or blood-testes sanctuary sites (Table 1). Whereas induction-related response to treatment has long been associated with EFS, the importance of minimal residual disease (MRD) as a biomarker in determining risk group stratification has been a relatively recent advance in ALL management.4,6,20 In the current treatment era, MRD-based risk assignment is being used to determine treatment for numerous leukemia trials. The The Associazione Italiana di Ematologia Oncologia Pediatrica and the Berlin-Frankfurt-Münster Acute Lymphoblastic Leukemia (AIEOP-BFM ALL 2000) study was the first to use PCR to assess immunoglobulin and TCR gene rearrangement MRD (PCR-MRD) at days 33 and 78 to risk-stratify 3184 patients to treatment.4 The COG is currently using surface antigen expression and flow cytometry at days 8 and 29 for risk assignment, and St Jude Children's Research Hospital continues to use flow cytometry to measure MRD for its treatment studies.21 Differences between PCR- and flow-based technologies and protocol-specific treatment approaches require continuous evaluation and optimization, especially as new molecular features are discovered that may carry prognostic importance.
Several molecular aberrations involving chromosomal aneuploidy, translocations, and point mutations have been described for pediatric ALL and together have become critical in defining high-risk subsets of patients. Based on an integrated approach using clinical features at presentation, response to treatment, and molecularly defined risk features, modern treatment algorithms have achieved OS rates in excess of 80%. Nevertheless, patients with high-risk disease still remain at increased risk for relapse and treatment-related complications from intensified therapy and often suffer from long-term side effects.
The ephemeral nature of assessing risk for relapse in ALL
Despite the use of multiple, independent prognostic features to assess chance for relapse, approximately half of patients who relapse are those found to have favorable clinical features and an excellent response to induction therapy.22 The emergence of resistant clones poses a difficult challenge regardless of the previous treatment intensity.23,24 It is possible that the size of a resistant subclone may determine whether a patient fails induction or has an early relapse, and also may govern the ease of identification of an underlying high-risk molecular feature.24 As a further complication, the drugs used to treat ALL have remain largely unchanged over the last several decades, so patients with relapsed disease are essentially treated with the same drugs, albeit with greater intensity. In the setting in which drug-effluxing transport proteins have been up-regulated, especially for ABCB1 (MDR1) and ABCC1 (MRP1), multidrug resistance to conventional chemotherapy can be especially difficult to overcome.25,26 The search for molecularly defined risk factors and targeted therapies stands at the forefront of translational research in pediatric ALL. Therapies directed to a specific signaling pathway should ideally include the identification of a driving molecular aberration that, in a leukemic clone, controls its growth and proliferation (“oncogene addiction”) and can be specifically disrupted by a small molecule having little or no toxicity. As discussed in the article by Dr Hunger, “Tyrosine Kinase Inhibitor Use in Pediatric Philadelphia Chromosome–Positive Acute Lymphoblastic Leukemia,” such a paradigm exists as a model approach for children with ALL having the Philadelphia chromosome translocation (Table 2 and reviewed by Hunger27 ).
Driving molecular aberrations and kinase signaling in pediatric precursor-B ALL
Protein kinases are key regulators of cell function and constitute one of the largest and structurally diverse groups of genes. Kinases function by transferring phosphate groups to acceptor proteins, and in so doing, direct the function of almost all cellular activities, including cell division, proliferation, and differentiation. As detailed elsewhere for BCR-ABL, molecular translocations that result in uncontrolled kinase signaling have been identified in several genetically defined ALL subsets. In 2001, a novel receptor subunit was described for the thymic stromal lymphopoietin28,29 and termed cytokine receptor-like factor 2 (CRLF2; Table 2). In 2009, Russell et al30 reported novel, cryptic translocations involving CRLF2: t(×;14)(p22;q32) or t(Y;14)(p11;q32)30 or deletions involving the pseudoautosomal region (PAR1), either del(×)(p22.33) or del(Y)(p11.32p11.32) that occur in 5% of childhood ALL. Overexpression of CRLF2 was associated with activation of the JAK-STAT pathway and proliferation in B-cell progenitors, suggesting a causal role in lymphoid transformation. Interestingly, many patients with Down syndrome have been found to have CRLF2 mutations, yet without a worse chance for cure.31 In primary patient samples, deregulation of CRLF2 was associated with mutations of JAK2 (different from the V617F mutation commonly seen in myeloproliferative disorders). Subsequently, in a genome-wide analysis of 207 patients with National Cancer Institute (NCI) high-risk features who were treated in the COG P9906 and 1961 studies, Harvey et al found a cohort in which overexpression of CRLF2, concurrent deletions of IKZF1, and other aberrations were associated with an EFS of approximately 15%-20% and a gene-expression signature reflective of activated tyrosine kinases (Figure 2).32,33

Heat-map analysis in 206 children and young adults with high-risk precursor B-ALL treated in COG study 9906. The R1 cohort captured all patients with MLL rearrangements and the R2 cohort captured all patients with the t(1;19). The R8 cohort was found to have BCR-ABL–like signature,32 CRLF2 mutations, and a poor outcome. Other mutations involving JAK were identified, suggesting that this cohort might respond to tyrosine kinase–directed therapy. The remaining R cohorts have molecular anomalies that remain undefined. (Used with permission from Harvey et al.33 )
Heat-map analysis in 206 children and young adults with high-risk precursor B-ALL treated in COG study 9906. The R1 cohort captured all patients with MLL rearrangements and the R2 cohort captured all patients with the t(1;19). The R8 cohort was found to have BCR-ABL–like signature,32 CRLF2 mutations, and a poor outcome. Other mutations involving JAK were identified, suggesting that this cohort might respond to tyrosine kinase–directed therapy. The remaining R cohorts have molecular anomalies that remain undefined. (Used with permission from Harvey et al.33 )
Others have shown that CRLF2 mutations either did34 or did not32 portend a worse prognosis, suggesting that patient selection and treatment may play important roles in the efficiency of this molecular marker. Several hematologic malignancies appear to be driven by molecular aberrations that affect the JAK-STAT pathway, including mutations of JAK2 and JAK1,35,36 leading to the targeted development of small molecules that might control de-regulated signaling. In aggregate, these and other gene abnormalities, including deletions or mutations of Ikaros zinc finger 1 (IKZF1), which appears to be causally related to leukemogenesis in animal models, may result in an inferior EFS in human ALL.36 Because of the association between CRLF2 overexpression and JAK mutations, patients with refractory/relapsed leukemia with known mutations of CRLF2 and/or JAK are eligible for enrollment in COG study ADVL1011 to receive a JAK inhibitor, INCB018424 (Ruxolitinib; Incyte Corporation).
Novel therapies for other kinase-dependent signaling pathways
The PI3K-akt and Ras-MAPK prosurvival signaling pathways are required for normal homeostasis in nonmalignant cells, but it is a continued reliance or “addiction” by leukemia cells on these pathways that has made them popular for targeted therapies. The PI3K-Akt pathway is often constitutively up-regulated in many lymphoid malignancies (reviewed by Lee-Sherick et al37 ), leading to the development of targeted therapies against the mammalian target of rapamycin (mTOR) and the TOR complexes.38 The inhibition of mTOR has been found to be effective in pediatric ALL,39,40 leading to the evaluation of temsirolimus (CC1-779) in phase 1 clinical trials. Although specific driving molecular aberrations have yet to be identified for ALL cells that are reliant on the Aurora serine/threonine, RAS/RAF/ERK, and Tyro-3, Acl, and Mer (TAM) families of kinases, therapies targeted against them are in various stages of development. Currently, a phase 1/2 trial is under way for MLN8237, an Aurora kinase inhibitor. Sorafenib, a tyrosine kinase inhibitor having a broad range of targets, is in phase 1 investigation for several malignancies, including pediatric ALL. A small-molecule inhibitor of Mer is being investigated for its efficacy in pediatric ALL cell lines.37
Driving molecular aberrations in pediatric T-lineage ALL
T-ALL is a biologically distinct disease entity that is associated with clinically aggressive features and poor treatment outcomes. Numerous studies have shown that, compared with patients with precursor B-ALL, children with T-ALL are more likely to present with NCI high-risk features, including a high WBC (> 50 000 cells/μL), age ≥ 10, and leukemic infiltrates into the CNS (Table 1).41,42 Adjusted for NCI-designated clinical risk factors, patient outcome in T-ALL remains inferior to EFS rates achieved for pre-B ALL. Whereas the NCI risk factors indicating high-risk disease have been relatively unhelpful in T-ALL, the detection of measurable MRD levels has become increasingly established as a means of identifying patients who are at higher risk for relapse.6,20,22,27
The early T-Precursor phenotype and its therapeutic implications
Recently, Coustan-Smith et al hypothesized that T-lymphoblasts with stem-cell-like features might respond less favorably to ALL-directed therapies.43 Working with T-ALL samples banked at Jude Children's Research Hospital and the AIEOP ALL-2000, 30 samples were identified to have early T-precursor (ETP) antigen profiles that were characterized by CD1a(−), CD8(−), CD5(weak) with stem-cell or myeloid markers (Table 1). This patient cohort also showed increased size and number of gene lesions, suggestive of an increased genomic instability. Compared with a 10-year relapse rate of only 10% for other T-ALL patients, those with ETP had a 75% relapse rate. A recent analysis of ETP patients registered in COG AALL0434 showed that 74% had end-induction MRD levels that were > 1%, a feature previously associated with a high chance of relapse in earlier BFM-based approaches.20
A search for specific, driving molecular aberrations in ETP patients has raised the possibility that an absence of biallelic TCRγ locus deletions (ABD) was predictive of induction failure in Dana-Farber and COG cell-bank registries. Using Chromosome Conformation Capture on a Chip (4C) technology, mutations of myocyte-specific enhancer factor 2C (MEF2C) were associated with the ETP profile and had a similarly poor outcome.44 Building on work done by St Jude Children's Research Hospital and the iBFM, Mullighan and Shochat have recently reported several additional gene mutations in ETP patients, as well as newly discovered mutations of the IL-7 receptor that did not confer a worse outcome.45 Many ETP patients fail to achieve a first remission, which may be mediated by a G0G1 cell-cycle arrest (Figure 3), protecting such cells from the cytotoxic drugs commonly used during induction.26 These findings suggest that the ETP signature may be a heterogeneous group of mutations and molecular rearrangements that result in an immature T-cell phenotype, which subsequently confers an intrinsic resistance to conventional chemotherapy approaches.

Pathway analysis of patients for whom induction failed in COG study 9404. The analysis shows arrest at G0/G1, suggesting that affected cells are not proliferating rapidly, thus conferring an inherent resistance to the cell-cycle therapies that are given during induction therapy. In these patient samples, subsequent analysis also showed an up-regulation of the MDR1 and MRP1 genes, which code for P-glycoproteins that are well known to confer chemoresistance in a variety of hematological malignancies. (Used with permission from Winter et al.26 )
Pathway analysis of patients for whom induction failed in COG study 9404. The analysis shows arrest at G0/G1, suggesting that affected cells are not proliferating rapidly, thus conferring an inherent resistance to the cell-cycle therapies that are given during induction therapy. In these patient samples, subsequent analysis also showed an up-regulation of the MDR1 and MRP1 genes, which code for P-glycoproteins that are well known to confer chemoresistance in a variety of hematological malignancies. (Used with permission from Winter et al.26 )
Whereas specific, driving molecular abnormalities remain to be defined for high-risk T-ALL, several therapeutic candidates for high-risk disease are being investigated in current clinical trials. In reports from the COG and CALGB, nearly 50% of children and 28% of adults with relapsed or refractory T-ALL46,47 achieved a second complete remission with nelarabine, even when used as a single agent. Nelarabine is currently being investigated for its efficacy in T-ALL in the context of a randomized clinical trial, COG AALL0434. In patients with relapsed T-ALL, the proteosomal inhibitor bortezomib is currently being used in conjunction with conventional reinduction therapy in the ADVL07P1 COG study. Although this study is still accruing patients and requires further follow-up, early data for the use of bortezomib in T-ALL have been encouraging, prompting interest for its inclusion in future clinical trials. Almost half of all T-ALL cases harbor mutations of NOTCH1. Interestingly, NOTCH1 mutations do not appear to confer either a good or bad risk status, but because of the high frequency of NOTCH1 mutations in T-ALL, it has become a focus of interest for targeted therapies (reviewed by Lee-Sherick37 ).
Future considerations for drug development in high-risk pediatric leukemia
Over the preceding 5 decades, marked improvement in survival rates for pediatric AML and ALL have been achieved through the use of intensified cytotoxic therapy. Current dose-intensified therapies, however, are limited by several serious acute and long-term toxicities that call for comprehensive supportive care. The inclusion of molecularly targeted agents has necessitated a reevaluation of the drug-development paradigm. Important issues to be considered in the development of molecularly targeted agents are: (1) the stringency of testing of a particular biomarker during its development48 ; (2) determining which patients should receive a novel agent based on the presence of a biomarker; and (3) finding out whether the drug should be administered alone or in conjunction with other drugs.49
In a realistic setting, gene markers may be somewhat imprecise. For example, mutations in a rearranged tyrosine kinase receptor gene may mitigate against the effects of a specific tyrosine kinase inhibitor (eg, resistance to imatinib in certain Philadelphia chromosome–positive ALL patients) or more than one driving molecular aberration may be seen in high-risk precursor-B ALL (CRLF2, JAK, and IKZF) and T-ALL (ABD and/or ME2KC). Further, expression profiles for high-risk disease may not be accurate when drawn from samples with low viability during preprocessing, with hybridization artifacts, or with normalization problems.26 In such instances, care must be taken to avoid nonrandomly assigning patients to treatments based on a molecular aberration that is low in prevalence or has not be rigorously tested for sensitivity and specificity. Because few molecular biomarkers have met these conditions, the selection of patients who might be assigned treatment based on the finding of molecular abnormality must be carefully monitored to ensure safety. Similarly, targeted therapies may be more effective when used in the setting of conventional therapy, but must be used in phases of therapy when their effects would not interact with other treatments. In Figure 4, 4 scenarios are described in which a clinical trial might be designed in the absence of a biomarker or with a biomarker that is in various stages of testing and validation for treatment effect in conjunction with a targeted therapy.

Clinical trials design using a biomarker. In the first scenario, no biomarker is used in a randomized clinical trial. With enough patients, a 2 × 2 design may be used. In the second scenario, a biomarker that is being tested for specificity and sensitivity is being tested for treatment effects in experimental and control groups. In the third setting, patients are randomized to either test or not test a promising biomarker. Those with the biomarker are nonrandomly assigned to a specific treatment approach. Those without the biomarker receive conventional therapy. In the fourth scenario, a validated biomarker is being used to nonrandomly assign treatment. It is assumed in the fourth scenario that the biomarker has high sensitivity, specificity, and prevalence within the treatment population to merit its use to nonrandomly assign therapy. In the comparative, conventional therapy group, it is assumed that the therapy has not been significantly modified from that given to earlier patient subsets being used as historical controls. (Adapted from Young et al.48 )
Clinical trials design using a biomarker. In the first scenario, no biomarker is used in a randomized clinical trial. With enough patients, a 2 × 2 design may be used. In the second scenario, a biomarker that is being tested for specificity and sensitivity is being tested for treatment effects in experimental and control groups. In the third setting, patients are randomized to either test or not test a promising biomarker. Those with the biomarker are nonrandomly assigned to a specific treatment approach. Those without the biomarker receive conventional therapy. In the fourth scenario, a validated biomarker is being used to nonrandomly assign treatment. It is assumed in the fourth scenario that the biomarker has high sensitivity, specificity, and prevalence within the treatment population to merit its use to nonrandomly assign therapy. In the comparative, conventional therapy group, it is assumed that the therapy has not been significantly modified from that given to earlier patient subsets being used as historical controls. (Adapted from Young et al.48 )
Because of their very nature, pediatric acute leukemias are associated with immune compromise. In particular, patients with relapsed disease or very intensified initial therapy are at particular risk for the development of opportunistic infections or bleeding complications. In testing targeted drugs in such patients, supportive care measures to prevent against treatment toxicities are of particular importance. In particular, the COG is testing several supportive care measures within its Cancer Control (CCL) portfolio to include the evaluation of antifungal protection in AML patients (ACCL0933), the prevention of treatment-related weight loss (ACCL0935), acupressure to control chemotherapy-induced nausea and vomiting (ACCL1032), and other relevant topics. These important studies, in conjunction with other efforts to minimize the burden of modern treatment, will allow targeted therapies to be explored to their fullest extent in high-risk pediatric leukemias.
Acknowledgments
With great thanks to the Cancer Therapeutic Evaluation Program for its support of pediatric therapeutic clinical trials, to the Children's Oncology Group and its respective committees and members, and to the American Society of Hematology Education Committee.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Stuart S. Winter, Division of Pediatric Hematology/Oncology, MSC10 5590, 1 University of New Mexico, Albuquerque, NM 87131-5311; Phone: (505) 272-4461; Fax: (505) 272-8699; e-mail: swinter@salud.unm.edu.
