
Abstract
There is no single category in the fourth edition (2008) of the World Health Organization (WHO) classification of myeloid neoplasms that encompasses all of the diseases referred to by some authors as the myeloproliferative neoplasm (MPN) “variants.” Instead, they are considered as distinct entities and are distributed among various subgroups of myeloid neoplasms in the classification scheme. These relatively uncommon neoplasms do not meet the criteria for any so-called “classical” MPN (chronic myelogenous leukemia, polycythemia vera, primary myelofibrosis, or essential thrombocythemia) and, although some exhibit myelodysplasia, none meets the criteria for any myelodysplastic syndrome (MDS). They are a diverse group of neoplasms ranging from fairly well-characterized disorders such as chronic myelomonocytic leukemia to rare and thus poorly characterized disorders such as chronic neutrophilic leukemia. Recently, however, there has been a surge of information regarding the genetic infrastructure of neoplastic cells in the MPN variants, allowing some to be molecularly defined. Nevertheless, in most cases, correlation of clinical, genetic, and morphologic findings is required for diagnosis and classification. The fourth edition of the WHO classification provides a framework to incorporate those neoplasms in which a genetic abnormality is a major defining criterion of the disease, such as those associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, and FGFR1, as well as for those in which no specific genetic defect has yet been discovered and which remain clinically and pathologically defined. An understanding of the clinical, morphologic, and genetic features of the MPN variants will facilitate their diagnosis.
Introduction: WHO classification of MPN variants
The myeloproliferative neoplasm (MPN) variants are relatively uncommon myeloid neoplasms characterized by the proliferation and maturation of one or more of the myeloid lineages, are BCR-ABL1−, and do not meet the diagnostic criteria for the other BCR-ABL1− “classical” MPNs, polycythemia vera, essential thrombocythemia, and primary myelofibrosis.1–3 They also do not meet the criteria for any myelodysplastic syndrome (MDS), although they may have dysplastic as well as myeloproliferative features. The disorders usually included are chronic eosinophilic leukemia (CEL) and related entities, chronic neutrophilic leukemia (CNL), systemic mastocytosis (SM), the myelodysplastic/myeloproliferative neoplasms (MDS/MPN), chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML), atypical chronic myeloid leukemia, BCR-ABL1− (aCML), and a “provisional” entity, refractory anemia with ring sideroblasts with marked thrombocytosis (RARS-T).1,3,4
There is not a World Health Organization (WHO) category that encompasses all MPN variants; instead, they are distributed among 3 major subgroups of myeloid neoplasms: MPN, MDS/MPN, and myeloid and lymphoid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGRB, or FGFR1 (Table 1). To understand the rationale for the current categorization of these neoplasms, it may be helpful to consider the basic principle of the WHO classification. The classification uses all available information—clinical, morphologic, genetic, immunophenotype, and other biologic features—in an attempt to define clinically significant disease entities, although the relative contribution of any one of these parameters to the final diagnosis varies according to the disease.5,6 As the focus in myeloid neoplasms turns increasingly to the genetic infrastructure of neoplastic cells, and particularly to molecular abnormalities that may be targets for therapy, it is expected that genetic and molecular data will be increasingly incorporated into diagnostic algorithms and/or the nomenclature for myeloid malignancies. When the 2008 WHO classification was formulated, several genetic abnormalities were recognized to be closely associated with subgroups of myeloid neoplasms or with specific disease entities, and many of these were incorporated into the classification. For MPNs and MPN variants, the discovery of rearrangements or mutations of genes encoding protein tyrosine kinases involved in signal transduction not only contributed to an improved understanding of their pathogenesis, but also allowed for better diagnostic tools and improved classification. In some instances, such as neoplastic eosinophilia with rearrangements of PDGFRA, PDGFRB, or FGFR1, the genetic defect became the major criterion for naming the disease, and in cases involving PDGFRA or PDGFRB, for identifying a specific target for therapy. In other instances, such as the BCR-ABL1− MPNs that are often associated with the JAK2 V617F mutation, the genetic abnormality provides an objective criterion that identifies the myeloproliferation as neoplastic rather than reactive, as does mutated KIT in SM.
The 2008 WHO classification for myeloid neoplasms

*These are referred to as “MPN variants” by some authors.
†For a complete listing of MDS and AML subcategories, see Vardiman et al.6
Since the publication of the WHO classification in 2008, several additional genetic abnormalities have been discovered in the MPN variants, including mutations of RUNX1, TET2, CBL, ASXL1, EZH2 and IDH1/IDH2, some of which will likely be incorporated into diagnostic algorithms in the future. However, with the possible exception of the neoplasms associated with rearrangement of PDGFRA, PDGFRB, or FGFR1, no currently recognized genetic or molecular defect is entirely specific for any MPN or MPN variant. Therefore, to classify a myeloid neoplasm according to the WHO scheme, a multidisciplinary approach using multiple disease parameters is required.
Evaluation of patients with MPN variants
At times, the diagnosis of an MPN variant can be challenging. It may be difficult, for example, to distinguish cases of CNL, CEL, or CMML from several underlying illnesses that result in reactive neutrophilia, eosinophilia, or monocytosis, respectively. In other cases, a confusing mixture of myeloproliferative and myelodysplastic features can lead to problems in classification, and in yet others, the distinction between acute myeloid leukemia (AML) and an MPN variant, particularly CMML with abnormal monocytes, can be exceedingly difficult. The evaluation of patients suspected to have an MPN variant should follow guidelines similar to those for any other myeloid neoplasm, and should be directed toward ultimate correlation of clinical, morphologic, genetic, and other data relevant to establishing the diagnosis. Nevertheless, some specific issues are important to emphasize when considering this group of neoplasms. First, morphology is a key criterion in the diagnosis of all MPN variants, even for those associated with specific genetic defects; if peripheral blood, BM aspirate, and biopsy specimens are not of sufficient quality to allow a diagnosis, repeat specimens should be obtained. Second, the diagnosis should be based on specimens obtained before any definitive therapy for the myeloid proliferation. Therapeutic agents, including growth factors, can alter morphology and the proliferative aspects of a case and result in an incorrect diagnosis.6,7 Third, the diagnosis of MDS/MPN is applicable only for cases that initially have both myelodysplastic and myeloproliferative features and not to cases of MPN that later acquire dysplasia due to progressive disease or therapy. Fourth, the blast percentage is a useful tool for diagnosis and for predicting prognosis in myeloid neoplasms and is best obtained by visual inspection of blood and cellular BM aspirate smears. The peripheral blood of patients with AML with monocytic differentiation may appear deceptively similar to those of MDS/MPN subtypes with monocytosis, specifically JMML and CMML, so it is important to investigate the BM in all cases. In the WHO classification, promonocytes in the blood and BM are considered as “blast equivalents” when tallying the blast percentage. Their recognition is essential to distinguishing AML from CMML and JMML, but there is poor reproducibility among many hematologists and pathologists in recognizing promonocytes and distinguishing them from the more mature but frequently atypical monocytes often observed in CMML.7,8 Fifth, flow cytometry studies may provide supporting evidence for the diagnosis of some MPN variants. Even when not substantially increased in number, the blasts of patients with MPN or MDS/MPN may have phenotypic abnormalities such as expression of CD7 and CD56, overexpression of CD34 and CD15, or partial loss of CD13, CD33, and CD117. When 2 or more such abnormalities are present, they argue in favor of a neoplastic disorder.9 Similarly, the finding of 2 or more phenotypic abnormalities of monocytes, such as expression of CD56 and/or CD2 or underexpression of HLA-DR, can provide supportive evidence for the diagnosis of CMML when there are no cytogenetic abnormalities and/or minimal dysplasia.7,10 Finally, a complete karyotypic analysis is essential at the time of workup of any myeloid disorder, and further FISH and molecular testing should be performed according to the clinical and morphologic findings of the individual case. Because of the various clinical manifestations of chronic myelogenous leukemia (CML), BCR-ABL1+, cytogenetic, FISH, and/or molecular studies are necessary to exclude CML whenever CMML, JMML, CNL or CEL are considered. In the case of persistent eosinophilia for which no apparent cause is found, cytogenetic studies and FISH or molecular studies for PDGFRA, PDGFRB or FGFR1 rearrangements should be performed. Mutational analysis for aberrations that help to confirm a specific diagnosis for any of the MPN variants, such as NRAS, KRAS, PTPN11, NF1, or CBL mutations in JMML or JAK2 V617F in RARS-T, or that may contribute significant prognostic information should be considered in the specific context of the diagnosis being considered.7
Specific MPN variants: rationale and criteria for diagnosis and classification and genetic abnormalities
Myeloid/lymphoid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1 and CEL, NOS
The 2008 WHO classification uses a “semi-molecular” approach to classify myeloid neoplasms in which eosinophils are a predominant component. The rare neoplasms that fall into the subgroup, myeloid/lymphoid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1, are caused by the translocation of one of these genes to any one of several partner genes to form an abnormal fusion gene that results in constitutive activation of a receptor tyrosine kinase.11–14 In the case of the PDGFRA abnormality, the most common rearrangement is the FIP1L1-PDGRA fusion formed by an interstitial cryptic deletion at 4q12. Most cases with rearranged PDGFRA have persistent eosinophilia, often accompanied by mast cell proliferation. Although the mast cells may aberrantly express CD2 and/or CD25, as is commonly observed in SM, they may not form the cohesive clusters usually seen in SM.13 Furthermore, rearranged PDGFRA and mutated KIT are reportedly mutually exclusive, so mast cell proliferation with rearranged PDGFRA does not carry mutated KIT.14 Occasionally, patients with rearranged PDGFRA may present with T-lymphoblastic leukemia with eosinophilia.15 Patients whose leukemic cells have PDGFRB rearrangements often have a leukemic picture resembling CMML with eosinophilia or CEL; presentation as a lymphoblastic neoplasm has not been reported to date. The most common rearrangement of PDGFRB is t(5;12)(q31-33;p12);ETV6-PDGFRB, but more than 20 different partner genes have been identified and are associated with an abnormal karyotype. The hematopoietic neoplasms with FGFR1 rearrangements are heterogeneous. The initial presentation may be as MPN with eosinophilia, (“8p11 myeloproliferative syndrome”), but presentation as T-lymphoblastic leukemia/lymphoma is nearly as common. Furthermore, patients who present initially with a myeloid neoplasm may undergo transformation to T- or B-lymphoblastic leukemia or to AML, whereas patients with lymphoblastic disease may also transform to AML.16 The most common translocation fuses FGFR1 at chromosome 8p11 and ZNF198 at chromosome 13q12, but several variants have been reported.
The finding of rearranged PDGFRA or FGFR1 in both myeloid and lymphoid neoplasms suggests bilineal differentiation from an affected pluripotent stem cell.13,17 However, a subset of T cells normally expresses FGFR1, and constitutive activation of the receptor due to the rearrangement in a precursor cell may lead to autonomous expansion.18
It might be argued that this group of neoplasms would be more appropriately classified according to their initial presentation—for example, CEL, CMML with eosinophilia or lymphoblastic lymphoma/leukemia with eosinophilia—but overall this subcategory exemplifies the WHO principles by combining morphologic and genetic features to define the disease and, in the cases with PDGFRA and PDGFRB abnormalities, even to identify the target for anti-tyrosine kinase inhibitor therapy. What more could we expect from a classification?
For patients with eosinophilia in whom there is no abnormality of PDGFRA, PDGFRB, or FGFR1, the diagnosis of CEL, NOS may be possible (Table 2). For persistent eosinophilia (> 1.5 × 109/L) that fails to meet criteria for any WHO-defined category of neoplastic eosinophilia, that lacks a BCR-ABL1 fusion gene or an AML-associated recurrent genetic abnormality, and for which no reactive cause can be found, the diagnosis of idiopathic hypereosinophilia or, if organ damage is present, idiopathic hypereosinophilic syndrome is appropriate.14

SM
SM is included in the MPN category (Table 1). It is characterized by a proliferation of morphologically and immunophenotypically abnormal mast cells occurring in multifocal, compact cell clusters in one or more extracutaneous organs19 (Table 3). SM is almost invariably demonstrated in BM biopsy specimens, but any tissue can be infiltrated. Systemic mastocytosis is further subcategorized as indolent SM, aggressive SM, SM associated with another clonal hematologic non-MC-lineage disease, and mast-cell leukemia.19

SM is further subclassified as indolent mastocytosis, SM with associated clonal hematological non-mast-cell lineage disease, aggressive mastocytosis, mast cell leukemia, mast cell sarcoma, and extracutaneous mastocytoma. Cytogenetics: no specific abnormality; ∼30% reported to have clonal abnormalities, del(20q), del(11q) is the most frequently reported.46 Patients with SM with associated clonal hematological non-mast-cell lineage disease may exhibit cytogenetic abnormality of the associated neoplasm. Molecular genetics: most cases have the KIT D816V mutation, but detection depends on the sensitivity of the method and the number of neoplastic cells in the specimen. Mutations of TET2 occur in ∼30% of SM often associated with monocytosis with or without mutated KIT. Less frequent mutations include JAK2 V617 and N-RAS.22
At times, the mast cell infiltrate may be subtle or obscured by the fibrosis that often accompanies the infiltrate, particularly in the BM. The recent availability of reagents for the immunophenotypic characterization of mast cells by flow cytometry and immunohistochemical techniques has considerably facilitated the diagnosis of SM in tissue biopsies and aspirates. Both normal and abnormal mast cells express mast-cell tryptase and CD117, but, in contrast to normal mast cells, neoplastic mast cells express CD25 and often CD2 as well.19,20 In addition, CD30 has been reported to be aberrantly expressed by mast cells in patients with aggressive SM, but not by normal mast cells and only rarely by the mast cells of patients with indolent SM.21 The genetic abnormality most frequently associated with SM is the gain-of-function mutation KIT D816V.22 The mast cells of most patients with SM carry KIT D816V, but its detection in clinical samples reportedly may vary from 50%-95% depending on the number of mast cells in the specimen and the sensitivity of the assay.22–25 Nearly 30% of patients with SM demonstrate mutated TET2, with or without demonstrably mutated KIT. Although mutated TET2 has been associated with monocytosis in SM, the mutation carries no proven prognostic influence and is not at all specific, because it is observed in other MPN and MDS/MPN entities.22,23 Cases of SM associated with eosinophilia should be investigated for PDGFRA rearrangement; such cases are not associated with mutated TET2 or KIT, and if rearranged PDGFRA is present, the case should be classified according to the genetic defect.23
CNL
Approximately 150 cases of CNL have been reported in the literature, but a recent in-depth review indicated that only 40 of these meet the current WHO criteria for CNL shown in Table 4.26 The major difficulty is to exclude reactive neutrophilia due to underlying inflammatory or neoplastic diseases, particularly plasma cell myeloma, that may abnormally produce G-CSF or similar cytokines. There are no known genetic abnormalities specific for CNL. Cytogenetic abnormalities are rarely described and include +8, +9, del(20q), and del(11q); JAK2 V617F has been reported in occasional cases.1,27,28 It is important to exclude cases of CML, BCR-ABL1+ in which the breakpoint in BCR is in the mu region, which results in the fusion protein, p230. Such cases, although associated with marked neutrophilia, are nevertheless CML.
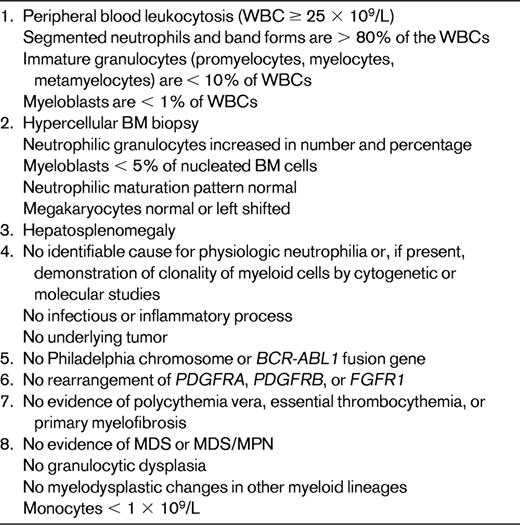
Very rare, most cases previously reported do not meet WHO criteria.26 Cytogenetics: no specific abnormality. Approximately 25%-30% of cases reportedly have clonal chromosomal abnormalities including del(20q), del(11q), or trisomy 8, 9, or 21.1,45,47 Molecular genetics: JAK2 V617F reported; incidence unknown.45
MDS/MPN
The MDS/MPN category encompasses those diseases that at the time of initial diagnosis have some clinical, laboratory, or morphologic features that support the diagnosis of MDS, such as persistent cytopenia(s) and dysplasia involving at least one myeloid lineage. They simultaneously exhibit other features, such as neutrophilia, monocytosis, thrombocytosis, or splenomegaly, that are more in keeping with an MPN. The entities included are shown in Table 1 and the diagnostic criteria for the most common MDS/MPN, CMML, are shown in Table 5. The specific entities CMML and RARS-T are discussed in the article by Drs Cazzola, Malcovati, and Inverneizzi, but general comments regarding the WHO classification of MDS/MPN are appropriate at this point.

Distinction of promonocytes from more mature but abnormal monocytes is key to distinguishing CMML from AML. Cytogenetics: clonal cytogenetic abnormalities in 20%-40%. The most frequent include +8, −7/del(7q), del(12p), and del(20q).40,44,48 Although most myeloid neoplasms associated with isolated isochromosome 17q will meet the criteria for CMML, others may be more appropriately categorized as MDS/MPN, unclassifiable. Some authorities argue that they are sufficiently unique to be considered as a separate entity.49 Molecular genetic: NRAS or KRAS, RUNX1, TET2, CBL, ASXL1 mutated in 20%-50%35,50–53 ; less commonly, EZH2 (11%-13%)42,53 ; IDH1/IDH2, JAK2, NPM1 (<10%)31,53,54 ; and, infrequently, FLT3, CEBP, WT1, and PTPN11 mutations.31,35
†Blasts include myeloblasts, monoblasts, and promonocytes.
This subcategory of myeloid neoplasms was introduced in the third edition of the WHO largely because several experts could not reach a consensus as to whether CMML and similar disorders such as aCML were myeloproliferative or myelodysplastic disorders. By the time the fourth edition of the classification was in preparation, the general notion that the pathogenesis of MPNs was often related to disturbances in tyrosine kinase signal transduction pathways, whereas abnormalities in the RAS signaling pathway seemed more frequent in MDS/MPN, were arguments to maintain the MDS/MPN category. More recently, the application of genetic techniques such as single nucleotide polymorphism arrays and high-throughput sequencing have revealed several genetic abnormalities, including mutations of genes encoding transcription factors such as RUNX1 and CEBPA—abnormalities rarely seen in MPNs—that buttress the argument that the MDS/MPN neoplasms are unique.29–31
There are no currently known cytogenetic or molecular genetic abnormalities specific for any MDS/MPN. The finding of BCR-ABL1 or rearrangements of PDGFRA, PDGFRB, or FGFR1 excludes the diagnosis of MDS/MPN, which are defined largely by their clinical and morphologic findings. In JMML, the finding of GM-CSF hypersensitivity of the leukemic cells has long been recognized as a hallmark finding, and led to the discovery of abnormalities in the GM-CSF receptor/RAS-RAF-MEK-ERK signal transduction pathway. In 80%-85% of cases of JMML, mutually exclusive somatic mutations of NF1, PTPN11, NRAS, KRAS, or CBL, lead to hyperactivation of the RAS pathway, are present and support the notion that constitutive activation of the RAS pathway is central to the pathogenesis.32–34 In the appropriate clinical setting, the discovery of one of these mutations can be used to support the diagnosis of JMML.
Several clinical and hematologic similarities between CMML and JMML, such as monocytosis, splenomegaly, and variable dysplasia, as well as the frequent finding of mutated NRAS and KRAS, initially led to the notion that they likely have a similar pathogenesis. More recent data, however, suggest some important differences. Mutated NRAS or KRAS is found in 25%-40% of cases of CMML, reportedly most often in patients in whom myeloproliferative rather than myelodysplastic features predominate.35 However, some data suggest that mutations in the RAS pathway in CMML are secondary abnormalities more important in disease progression than the initial pathogenesis.36 Although abnormalities of CBL have been reported in 5%-18% of cases of CMML,30,37 mutated NF1 or PTPN11 that occur in 40%-50% of cases of JMML are rare in CMML.38 In contrast, mutations affecting the transcription factors RUNX1, CEBPA, NPM1, or WT1 have been reported in up to 30% of cases of CMML, but to date not in JMML.31,38 Other mutations overrepresented in CMML, such as mutated JAK2, ASXL1, TET2, and IDH1/2, are also rare in JMML. The significance of these genetic abnormalities on the future classification of CMML or JMML is not clear, but the reported association of RAS mutations with the myeloproliferative features of CMML may provide an objective criterion for further subclassification into myeloproliferative-like and myelodysplastic-like subtypes, an issue addressed in a previous French-American-British (FAB) classification but not in the current WHO system.
Atypical CML earned a reputation as the worst-named disease in the third edition of the WHO classification because the name implies it is merely CML with atypical features. Therefore, the name, which was initially adopted from the FAB system, was changed in the fourth edition to “atypical CML, BCR-ABL1−” to emphasize that it is distinctly different from CML, BCR-ABL1+.39 The criteria and genetic features of aCML are listed in Table 6. The hallmark of aCML is marked granulocytic dysplasia, but other lineages may also be dysplastic. Morphologically, aCML may be difficult to distinguish from CMML; if a monocytic component is detected but does not meet the level required for a diagnosis of CMML, it is prudent to provide a differential diagnosis and reassess after an appropriate interval of time. Although 80% of cases of aCML have cytogenetic abnormalities, most commonly +8 or del(20q), the abnormalities are not specific.40 There are similarities between aCML and CMML at the molecular level. Although NRAS and KRAS mutations occur in aCML, the JAK2 V617F, found in nearly 10% of cases of CMML, is rarely if ever seen in aCML.41 However, mutations of CBL, RUNX1, CEBPA, EZH2, and WT1 occur in a similar frequency as in CMML.31,42

Difficult at times to distinguish from CMML, but granulocytic dysplasia is usually much more severe than in CMML. Nonspecific esterase staining of BM samples may be helpful in demonstrating the number of monocytes. Cytogenetics: clonal cytogenetic abnormalities in up to 80%; trisomy 8 and del(20q) are the most common, but abnormalities of chromosomes 13, 14, 17,19, and 12 are also commonly reported.40 Molecular genetics: mutated NRAS, KRAS, or TET2 in nearly 30%37 ; mutations of CBL, RUNX1, CEBPA, EZH2, or WT1 in 1%-10%31,42,55 ; and JAK2 V617F occurs rarely if at all.40
Summary
The WHO classification of the MPN variants requires correlation of clinical, morphologic, and genetic data for their proper categorization. Hopefully, in view of the rapidity with which the genetic infrastructure of the neoplastic cells is being determined in these diseases, future classifications will incorporate more molecular data to better define the neoplasm and possible therapeutic targets.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
James Vardiman, MD, Director, Hematopathology and Clinical Hematology Laboratory, The University of Chicago Medical Center, 5841 South Maryland Ave, Chicago, IL 60637; Phone: (773) 702-6196; Fax: (773) 702-1200; e-mail: james.vardiman@uchospitals.edu.
