
Abstract
Expansion of myeloid blasts with suppression of normal hematopoiesis is a hallmark of acute myeloid leukemia (AML). In contrast, myeloproliferative neoplasms (MPNs) are clonal disorders characterized by overproliferation of one or more lineages that retain the ability to differentiate. Juvenile myelomonocytic leukemia (JMML) is an aggressive MPN of childhood that is clinically characterized by the overproduction of monocytic cells that can infiltrate organs, including the spleen, liver, gastrointestinal tract, and lung. Major progress in understanding the pathogenesis of JMML has been achieved by mapping out the genetic lesions that occur in patients. The spectrum of mutations described thus far in JMML occur in genes that encode proteins that signal through the Ras/mitogen-activated protein kinase (MAPK) pathways, thus providing potential new opportunities for both diagnosis and therapy. These genes include NF1, NRAS, KRAS, PTPN11, and, most recently, CBL. While the current standard of care for patients with JMML relies on allogeneic hematopoietic stem-cell transplant, relapse is the most frequent cause of treatment failure. Rarely, spontaneous resolution of this disorder can occur but is unpredictable. This review is focused on the genetic abnormalities that occur in JMML, with particular attention to germ-line predisposition syndromes associated with the disorder. Current approaches to therapy are also discussed.
Introduction
Juvenile myelomonocytic leukemia (JMML) is an aggressive myeloproliferative neoplasm (MPN) of childhood that occurs with an estimated frequency of 1.2 cases per million.1 Because of the rarity of the disease and the heterogeneous nature of its clinical manifestations, the diagnosis of JMML is frequently challenging for the average general practitioner. Physicians are usually asked to consult on a young child with fever, splenomegaly, and a high white blood cell count with a peripheral monocytosis and a few circulating myeloid precursor cells, all of which are nonspecific features that can be associated with bacterial or viral infections. Indeed, one of the hallmark laboratory features of JMML, hypersensitivity of myeloid progenitor cells to granulocyte-macrophage colony stimulating factor (GM-CSF) in colony-forming assays,2 can also be present in certain viral infections, thus providing a sensitive but nonspecific adjunctive diagnostic tool. At presentation, a rare circulating nucleated red blood cell can also usually be found in the peripheral blood smear, and approximately 50% of patients will present with an elevated hemoglobin F corrected for age.1 Table 1 lists currently accepted and updated criteria for making the diagnosis of JMML.
Current diagnostic criteria for JMML
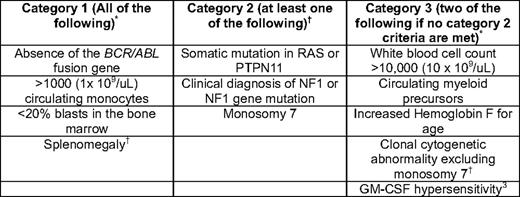
*Current WHO criteria.
†Proposed additions to the WHO criteria that were discussed by participants attending the JMML Symposium in Atlanta, GA, 2008.41 Note that CBL mutations have since been discovered and should be screened for in the workup of a patient with suspected JMML. Patients who are found to have a category 2 lesion need to meet criteria in category 1, but do not need to meet category 3 criteria. Patients who are not found to have a category 2 lesion must meet the category 1 and 3 criteria. Only 7% of patients with JMML will NOT present with splenomegaly, but virtually all patients develop this within several weeks to months of initial presentation (Niemeyer, personal communication).
Children affected by specific congenital syndromes are at a higher risk of developing JMML or a related transient myeloproliferative disorder in infancy, and the identification of genes associated with these syndromes have provided unique insights into the pathogenesis of this myeloid malignancy. Recent discoveries have advanced our ability to make a firm molecular diagnosis of JMML in 85% to 90% of children (Figure 1).1,3

Schematic diagram showing ligand-stimulated Ras activation, the Ras-MAPK pathway, and the gene mutations reported to date contributing to the neuro-cardio-facio-cutaneous congenital disorders, a newly defined Cbl germ-line syndrome, and JMML. NL/MGCL, Noonan-like/multiple giant cell lesion; CFC, cardia-facio-cutaneous. (Modified from Chan et al., 2009.41 Used with permission from Elsevier.)
Schematic diagram showing ligand-stimulated Ras activation, the Ras-MAPK pathway, and the gene mutations reported to date contributing to the neuro-cardio-facio-cutaneous congenital disorders, a newly defined Cbl germ-line syndrome, and JMML. NL/MGCL, Noonan-like/multiple giant cell lesion; CFC, cardia-facio-cutaneous. (Modified from Chan et al., 2009.41 Used with permission from Elsevier.)
Molecular Genetics Implicate Initiating Lesions in the Ras/Mitogen-Activated Protein Kinase Pathway
In 1978, it was noted that children affected by neurofibromatosis type 1 (NF1) were particularly prone to developing a “chronic myeloid leukemia” or acute myelomonocytic leukemia in the first decade of life.4 NF1 can be either an autosomal dominant disorder or can arise spontaneously. It is characterized by six or more “café au lait macules,” two or more neurofibromas or one plexiform neurofibroma, Lisch nodules, axillary or inguinal freckling, and/or optic gliomas.
In 1990, the NF1 gene was cloned and the encoded neurofibromin protein was identified to function as a GTPase activating protein for Ras, which greatly enhances the hydrolysis of the active, GTP-bound conformation of Ras to the inactive, GDP-bound conformation.5 In 1994, Shannon et al. demonstrated loss of the wild-type allele in the diseased bone marrow of children with JMML affected by NF1, thus establishing NF1 as a classic tumor suppressor gene.6 In subsequent mouse models engineered to conditionally delete the Nf1 allele, MPN develops and elevated levels of Ras-GTP occur, thus validating the hypothesis that the absence of normal neurofibromin results in a relative increase in Ras-GTP.7,8 In additional reports in 1994, Kalra and Miyauchi separately identified somatic, oncogenic mutations in NRAS or KRAS2 (RAS) occurring in a second, independent cohort of patients with JMML, thus providing additional genetic evidence to support the hypothesis that hyperactivation of the Ras/mitogen-activated protein kinases (MAPK) pathway was essential for the pathogenesis of this disease.9,10
Noonan syndrome is another congenital disorder that can either be inherited in an autosomal dominant fashion or arise spontaneously.11 It is characterized by facial dysmorphism, short stature, webbed neck, and cardiac anomalies, as well as varying levels of impaired cognition. Interestingly, some children with Noonan syndrome also display a hematologic phenotype, including a self-resolving myeloproliferative disorder in infancy that resembles JMML, and bleeding diatheses. In 2001, Tartaglia and Gelb identified germ-line mutations in PTPN11, a gene encoding the non-receptor tyrosine phosphatase protein SHP-2, as causative in approximately 50% of children with Noonan syndrome.12 Based on the observation that children with Noonan syndrome can display myeloproliferative features, mutations in PTPN11 were subsequently identified to occur as somatic lesions in de novo, non-syndromic JMML in as many as 35% of patients.13,14
The SHP2 phosphatase includes two Src homology 2 (SH2) domains (termed N-SH2 and C-SH2) and a catalytic protein tyrosine phosphatase (PTP) domain.15 Analysis of the crystal structure of SHP-2 revealed that the N-SH2 domain functions to directly block the active site of the PTP domain, thereby resulting in a catalytically inactive SHP-2 protein. The SHP-2 PTPase is activated when its SH-2 domains bind an appropriate tyrosine phosphorylated ligand, which results in a conformational shift that permits the active site to interact productively with target molecules. SHP-2 participates in signal transduction downstream of growth factor receptors to regulate multiple cellular responses, including proliferation, differentiation, and migration.
A comparison of the PTPN11 mutations in de novo versus syndromic JMML (Noonan syndrome) reveals that many, but not all, of the same codons in exons 3, 4, and 13 are affected.16 However, in Noonan syndrome the codon substitutions are largely different from those in JMML, leading investigators to hypothesize that the transforming ability of mutations in Noonan syndrome are “weaker” and thus may be tolerated as germ-line events. Indeed, the most common PTPN11 mutation in de novo JMML is 226G>A, resulting in E76K, an alteration that has never been documented as a germ-line lesion in Noonan syndrome. Studies of the E76K protein reveal that it displays the highest phosphatase activity, induces profound hypersensitivity to GM-CSF in fetal liver cells, and transforms transduced BaF3 cell lines to cytokine independence.14,17,18 The D61G mutation is the only one reported to occur in both Noonan syndrome and de novo JMML. Transgenic mouse models with this lesion develop a mild MPN with a latency of 5 months, in contrast to the D61Y, which is a lesion only occurring in JMML in which transplanted mice develop a fatal, JMML-like aggressive disease by 6 to 7 months.19,20
Recently, high-density single-nucleotide polymorphism arrays have revolutionized the ability to identify regions of copy number gain and loss in a variety of human cancers. Comparison of tumor tissue with germ-line also allows detection of copy-neutral loss of heterozygosity, otherwise known as acquired uniparental isodisomy. In cancer, this arises as a duplication of a region containing a mutation that frequently, but not always, behaves as a tumor suppressor gene, as has recently been shown to occur in cases of NF1-associated JMML.21,22
To determine if additional regions of uniparental isodisomy exist in JMML, we performed single-nucleotide polymorphism array analysis (Genome-Wide Human SNP Array 6.0, Affymetrix, Santa Clara, CA) on samples from 27 JMML patients with and without known PTPN11, RAS, or NF1 abnormalities.3 We identified a region of 11q uniparental disomy in five of these cases. Recent reports uncovered similar lesions in adults with atypical chronic myelogenous leukemia, chronic myelomonocytic leukemia, and myelodysplastic syndromes, and subsequent molecular analysis identified homozygous CBL mutations.23–25 All five of our patients also displayed homozygous CBL lesions. In collaboration with investigators from the European Working Group on Myelodysplastic Syndromes (EWOG-MDS), we subsequently detected a total of 27 CBL mutations in a group of 68 JMML patients with no other known lesions. The most common mutation is the c.1111T>C transversion resulting in a substitution of a histidine for a key tyrosine residue at codon 371 in the alpha-linker region of the protein. Others have since confirmed these findings.26,27
Cbl is an E3 ubiquitin ligase that is known to mark activated receptor and non-receptor tyrosine kinases and other proteins for degradation by ubiquitination, but also retains important adaptor functions.28 Most of the mutant Cbl proteins reported to occur thus far in myeloid malignancies result from missense mutations or splice site variants, and the majority have been shown to result in an encoded protein. We and others have shown that defective E3 ligase activity results from specific substitutions at this region (Y371H, Y371S) only in the absence of the wild-type protein.25,29 An initial review of the medical records of 21 children with JMML harboring these homozygous mutations revealed that a number of them also shared other phenotypic features, suggesting that these lesions might first occur as germ-line events. Indeed, further investigation of the germ-line tissues from 17 of these patients revealed heterozygous lesions, and additional studies of parental DNA, when available, indicated that these mutations are autosomally inherited in a dominant fashion approximately 50% of the time, while they arise spontaneously in the other 50%.29
Other mutations have been recently reported to occur rarely in JMML, such as AXSL1.30 In contrast to other MPNs of adulthood, JAK2 mutations do not occur in JMML. A single case report of a FLT3 mutation has been reported to date.31 Taken together, these data would suggest that the current spectrum of commonly occurring mutations in NF1, RAS, PTPN11, and CBL converge on the Ras/MAPK pathway in a highly specific way that remains to be fully elucidated.
Prognostic Factors in JMML
While the current standard of care for JMML is allogeneic hematopoietic stem-cell transplant (HSCT), continued controversy exists about identifying those patients who need to be moved quickly to HSCT versus those rare patients who might be observed. Certainly, we now anticipate that most patients with Noonan syndrome who present with a JMML-like MPN in the neonatal period will spontaneously resolve over the first year of life, somewhat akin to the transient myeloproliferative syndrome of Down syndrome (although Noonan syndrome-MPN is slower to resolve than Down syndrome-transient myeloproliferative disorder). Rare Noonan syndrome patients may require some low- or intermediate-dose chemotherapy to improve splenomegaly and to control high white blood cell counts, but there is no longer the expectation that these children will require HSCT if they show clinical improvement in the first year of life. In addition, unlike patients with Down syndrome, individuals with Noonan syndrome who suffer from transient MPN of infancy do not appear to be at an increased risk of myeloid malignancies later in childhood.
For patients without Noonan syndrome, there have been multiple case reports in the literature describing a phenomenon of “self-resolving” JMML. Matsuda et al. observed that three children with JMML bearing N- or KRASG12S demonstrated a milder clinical course with spontaneous clinical improvement.32 Flotho et al. were unable to confirm in the EWOG-MDS database that RASG12S was associated with long-term survival in the absence of HSCT.33 However, it was noted that among 12 patients observed without HSCT for more than 3 years from diagnosis, there were 5 patients experiencing long-term survival who harbored various RAS mutations. Of great interest in both series is that spontaneously resolving patients all presented with favorable prognostic factors: young age at diagnosis, platelet counts above 33,000/uL, and relatively low age-adjusted hemoglobin F levels.34,35
We now know that patients with homozygous CBL mutations have a high rate of spontaneous resolution, though this is not a uniform occurrence, as some children with CBL lesions have progressed and relapsed after HSCT. In addition, of 5 patients reported with spontaneously resolving JMML and the germ-line Cbl syndrome, 4 developed signs consistent with serious vasculitis by the end of their second decade of life, as evidenced by optic atrophy, hypertension, cardiomyopathy, or arteritis.29
As stated above, certain clinical factors have been associated with a poorer prognosis, and thus should sway investigators toward earlier HSCT. Such factors include older age at diagnosis (>2 years), increased fetal hemoglobin for age (Hgb F), and/or a platelet count under 33,000/uL at presentation, as well as female sex in some studies.34,35 These factors consistently correlate with a poor outcome (reduced event-free survival [EFS] and overall survival [OS]) following allogeneic stem cell transplantation.36
Several studies have also examined the hypothesis that mutational status may be correlated with JMML clinical features and prognosis; however, this remains controversial. In 2005, Locatelli reported improved EFS of 64% with a 3-year median follow-up for 100 patients treated with allogeneic bone marrow transplant in the EWOG-MDS consortium. In that series, mutational status of NF1, PTPN11, or RAS was not statistically significant as independent risk factors for survival. Yoshida et al. evaluated the clinical course and laboratory findings of 49 JMML patients, 32 of whom harbored mutations in NF1, KRAS, NRAS, or PTPN11.37 In their study, mutation of PTPN11 was associated with older age at diagnosis (>24 months), increased Hgb F (>10%), reduced overall survival, and, importantly, appeared to be an unfavorable prognostic factor predicting relapse following transplantation.
Recently, Bresolin et al. reported that gene expression signatures could segregate JMML patients into those who displayed an AML-type signature versus those who did not.38 These signatures were significantly associated with outcome, with those who displayed an AML-type signature experiencing a 10-year EFS of 6% and those without the AML-type signature who experience a 63% EFS. Of interest was the statistically significant correlation between an AML-type signature and known prognostic variables at diagnosis, including older age, lower platelet count, and higher Hgb F levels. However in a multivariate analysis, only the AML-type gene expression signature retained significance, although the levels of Hgb F and platelet count in this model were not defined at the same levels as previously determined to carry prognostic significance.
Taken together, the lack of prognostic significance of specific genetic mutations, as well as the lack of correlation between the AML-type signature and genetic lesions, suggests that additional, as-yet-unidentified factors are responsible for disease modulation and subsequent progression. Additional studies to identify these factors are ongoing.
Approaches to Therapy Prior to HSCT
Another area of controversy surrounding the care of JMML patients is the type of therapy delivered prior to HSCT. To date, no standard chemotherapy regimens used prior to HSCT have been shown to have an impact on the incidence of relapse post-HSCT.39 A wide variety of approaches have been used, from watchful waiting to high-dose AML-type chemotherapy. The Children's Oncology Group sponsored a phase II trial (AAML0122) that incorporated a window phase of a farnesyl-transferase inhibitor followed by cis-retinoic acid and cytarabine/fludarabine prior to HSCT. Clinical responses to the farnesyl-transferase inhibitor were encouraging; however, there were no data to suggest that inhibition of Ras prenylation accompanied these responses.40 Indeed, the promise of using farnesyl-transferase inhibitors as “Ras” inhibitors is misleading, because alternative geranylation can substitute for prenylation, a critical step in the activation of Ras. The EFS in the Children's Oncology Group trial was 40%, with an OS of 55% and a median follow-up of 4 years (Cooper, personal communication).
Similarly, the EWOG-MDS/EBMT JMML trial showed that patients who received either no pre-HSCT therapy or low-dose chemotherapy pre-HSCT compared with high-dose acute myeloblastic leukemia-like chemotherapy had identical EFS (52% vs. 50%), relapse incidence (35% vs. 38%), and treatment-related mortality (13% vs. 13%).36 Given the heterogeneity of disease burden at presentation in children with JMML, it is not possible to be definitive about the patients who were given various pretreatment therapies and the impact of those treatments on ultimate outcome.
Pretransplant splenectomy is also controversial. The previous Children's Oncology Group trial for JMML mandated splenectomy for all children prior to HSCT. However, the EWOG-MDS/EBMT JMML trial demonstrated that comparing children with a spleen size < 5 cm or > 5 cm, children undergoing splenectomy had no statistical benefit in event-free survival (EFS; 61% vs. 44% vs. 48%, respectively), relapse incidence (24% vs. 45% vs. 39%, respectively), or treatment-related mortality (15% vs. 11% vs. 13%, respectively).36 Therefore, neither splenectomy nor high-dose chemotherapy are recommended prior to a patient receiving conditioning for an allogeneic stem-cell transplantation unless clinically indicated for symptomatic relief.
There is growing consensus for watchful waiting for those patients who are asymptomatic while identifying an appropriate donor. For patients with very high white blood cell counts, pulmonary problems, and/or prominent organomegaly, one can consider oral 6-mercaptopurine (50 mg/m2/d) with or without cis-retinoic acid (100 mg/m2/d). For severely ill children, or those children who progress on less-intensive therapies, low-dose intravenous cytarabine (40 mg/m2/d for 5 d) can be administered. If this fails, the combination of high-dose cytarabine (2 g/m2/d for 5 d) plus fludarabine (30 mg/m2/d for 5 d) (both agents were dose adjusted for patients under 12 kg) can also be utilized and is generally well tolerated. To assess the efficacy of such therapies, clinical response criteria have been defined (Table 2).41 In addition to such criteria, allele-specific, polymerase chain reaction (PCR)-based strategies have been developed to track specific mutations in patients on therapy and will be tested in future clinical trials to determine their role prospectively.42
Proposed clinical response criteria for JMML*

*These are proposed response criteria only. Full current response criteria are listed in Chan et al., 2009.41
Future targeted therapies are being intensely investigated in a variety of laboratories around the country. Such potential therapies include Janus kinase (JAK) inhibitors, based on work demonstrating a pSTAT5 response in JMML cells upon exposure to sub-therapeutic concentrations of GM-CSF,43 as well as other signal transduction components of Ras signaling, such as PI3K (phosphoinositide 3-kinase), MEK (MAPK/extracellular-signal-regulated kinase [ERK] kinase), and mTOR (mammalian target of rapamycin) inhibitors.44
Approaches to HSCT
There is also controversy over current conditioning regimens prior to HSCT for JMML. Chemotherapy-based conditioning regimens, such as the one published by the EWOG-MDS, utilize busulfan, cytoxan, and melphalan, with reasonable transplant-related mortality and morbidity.36,45 The use of total body irradiation is now generally reserved for second transplants, and should be used cautiously in young patients, who may experience substantial late effects. Novel approaches to conditioning regimens are being developed that may incorporate strategies to further decrease late effects for JMML patients. Rapid taper of immunosuppression after HSCT should be initiated as quickly as possible, because a graft-versus-leukemia effect in JMML has been documented. In addition, novel methods of detecting relapse early post-transplant beyond traditional donor chimerism include sorted cell donor chimerism, quantitative PCR to assess chimerism, as well as allele-specific PCR strategies to detect mutation-specific clones.42,46 Such sensitive measurements may facilitate earlier and more rapid withdrawal of immunosuppression or the initiation of novel targeted therapies. There are minimal data on the utility of donor lymphocyte infusions.46,47
Finally, for those JMML patients who relapse after HSCT, there is growing experience with the use of second HSCT, with either the identical donor or an alternative donor. As many as 50% of patients with relapsed JMML can be salvaged with second transplant (Locatelli, unpublished data).
Summary and Directions for Future Work
In summary, JMML is a rare MPN of childhood in which a firm molecular diagnosis can now be made in approximately 90% of patients. Controversy continues to exist over the identification of rare patients who may be observed; however, the majority of patients require swift HSCT. Certain clinical factors may aid in that decision, such as older age, lower platelet count, and higher Hgb F levels at diagnosis. In addition, some patients with RAS or CBL mutations may experience spontaneous resolution, but it remains challenging to identify exactly who will do well without HSCT. Indeed, patients with CBL mutations who resolve spontaneously may develop serious complications later in life related to vasculitis, although further studies are needed in larger cohorts of patients to validate these early findings and to determine if patients with CBL mutations who have undergone HSCT are spared from developing this late complication. Finally, a stepwise approach to treating patients with JMML with chemotherapy prior to definitive HSCT can now be considered, because data remain elusive about the benefits of such chemotherapy prior to transplant.
Future studies will focus on refining prognostic risk factors and optimizing therapy for these patients. Ongoing investigations of targeted therapies will require continued study of the biochemical consequences of the genetic lesions occurring in JMML, and mandate fruitful collaborations between basic, translational, and clinical scientists.
Acknowledgments
MLL is supported in this work by the Leukemia Lymphoma Society (LLS) Translational Research Program (6059-09) and is a Clinical Scholar of the LLS.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests.
Off-label drug use: None disclosed.
Correspondence
Mignon L. Loh, MD, Department of Pediatrics, University of California, San Francisco, 513 Parnassus Ave., Rm. HSE-302, Box 0519, San Francisco, CA 94143; Phone: (415) 476-3831; e-mail: lohm@itsa.ucsf.edu
