
Abstract
Inflammation arising from various etiologies, including infection, autoimmune disorders, chronic diseases, and aging, can promote anemia. The anemia of inflammation (AI) is most often normocytic and normochromic and is usually mild. Characteristic changes in systemic iron handling, erythrocyte production, and erythrocyte life span all contribute to AI. The preferred treatment is directed at the underlying disease. However, when the inflammatory insult is intractable, or the cause has not been diagnosed, there are limited options for treatment of AI. Because anemia is a comorbid condition that is associated with poor outcomes in various chronic disease states, understanding its pathogenesis and developing new tools for its treatment should remain a priority. Hepcidin antimicrobial peptide has taken center stage in recent years as a potent modulator of iron availability. As the technology for quantitative hepcidin analysis improves, hepcidin's role in various disease states is also being revealed. Recent insights concerning the regulatory pathways that modify hepcidin expression have identified novel targets for drug development. As the field advances with such therapeutics, the analysis of the impact of normalized hemoglobin on disease outcomes will confirm whether anemia is a reversible independent contributor to the morbidity and mortality associated with inflammatory diseases.
Clinical Presentation
Anemia of inflammation (AI) can occur during infections with a microbial pathogen, including bacterial, viral, or yeast infections, or it can occur in the context of autoimmune disorders such as systemic lupus erythematosus or rheumatoid arthritis. AI may also result from chronic disease states with low-grade inflammatory activity, such as cancer, chronic kidney disease, or congestive heart failure. Even the pro-inflammatory state of aging, independent of disease, may result in anemia that is postulated to have a similar pathogenesis.
AI is most commonly described as a normocytic, normochromic anemia, but it can become microcytic and hypochromic as the disease progresses.1 Reticulocytosis is not usually observed. Characteristic changes in systemic iron distribution develop such that the serum iron concentration and transferrin saturation are low, while macrophage iron stores remain replete. Hypoferremia and sequestration of iron in tissue macrophages are thought to protect the host by limiting the availability of this micronutrient, which is required for the proliferation of most microbial pathogens.
A “blunted” erythropoietin (Epo) response has also been described. In some disease contexts, Epo production is not sufficiently increased relative to the severity of the anemia. Alternatively, the blunted Epo response has been described as the inability of the erythron to respond to available Epo. Finally, decreased erythrocyte life span has been reported in some patient groups with AI. These features of AI suggest that the immune response promotes iron sequestration, but also inhibits the production and survival of erythrocytes.2
While many of these features have been observed in a number of different patient populations, there is some variability in clinical expression that may be related to the duration or severity of the inflammation or to differential expression of various pro-inflammatory cytokines that direct the inflammatory response. It is conceivable that the variation in the “cytokine profile” across disease states may lead to variations in the mechanisms that restrict erythropoiesis or promote turnover of erythrocytes and therefore affect the presentation of AI.
Molecular Pathophysiology
The Normal Erythrocyte Life Cycle
Erythroid precursors at multiple developmental stages form physical interactions with a central macrophage (Figure 1A). This microenvironmental niche has been referred to as an “erythroid island.”3 Erythrocyte development proceeds in an Epo-dependent phase, which directs proliferation and survival of erythroid progenitors, followed by an Epo-independent phase, which is largely focused on hemoglobin production.4 Nucleated erythrocyte precursors called erythroblasts express high levels of transferrin receptor (TfR), which is required for iron uptake from serum diferric transferrin. When sufficient hemoglobin has been produced, the TfR is shed, forming soluble TfR (sTfR). The erythroblast then extrudes its nucleus and enters the circulation as a reticulocyte (Figure 1B). The reticulocyte continues to mature in the circulation as mitochondria are removed and mRNA levels decline. Eventually, the erythrocyte takes on the classic discoid shape and circulates in the blood for approximately 120 days in humans or 50 days in mice.

Erythroid precursors mature in the bone marrow in physical contact with a central macrophage (A). After completing hemoglobinization, erythroid precursors extrude their nucleus and enter the circulation (B). Senescent erythrocytes are phagocytosed by tissue macrophages, where the iron is recycled (C). Iron is returned to the erythron via diferric transferrin (D). (Figure courtesy of the author.)
Erythroid precursors mature in the bone marrow in physical contact with a central macrophage (A). After completing hemoglobinization, erythroid precursors extrude their nucleus and enter the circulation (B). Senescent erythrocytes are phagocytosed by tissue macrophages, where the iron is recycled (C). Iron is returned to the erythron via diferric transferrin (D). (Figure courtesy of the author.)
Senescent erythrocytes are phagocytosed by tissue macrophages, particularly those of the spleen (Figure 1C). Because dietary iron uptake alone is insufficient to meet the requirements of erythropoiesis, and because iron, heme, or hemoglobin excretion is extremely limited in the absence of pathology, the iron in hemoglobin must be recovered and recycled. Recently, new gene families have been identified that may participate in heme recycling. HRG-1 (heme responsive gene-1) localizes to intracellular compartments and facilitates cellular heme uptake.5 Feline leukemia virus, subgroup C, receptor (FLVCR) facilitates heme egress from erythroid precursors and macrophages.6 The quantitative contribution of extracellular heme to total iron fluxes is not known but is probably small. Most heme is degraded by heme oxygenase,7 and the iron can either be stored in ferritin within the macrophage or exported to the plasma (Figure 1D). Ferroportin (Fpn) is the only transporter known to facilitate elemental iron egress.2 Export from the macrophage as Fe2+ requires the activity of ceruloplasmin (Cp), a multi-copper oxidase, to convert Fe2+ to Fe3+ for efficient loading onto apo-Tf.8 The efficient turnover of iron from senescent erythrocytes constitutes the main source of iron for erythropoiesis and is critical to its maintenance.
Iron Sequestration
Low serum iron concentration is a hallmark of the clinical presentation of AI. In mouse models9 and in human acute inflammatory states,10 hypoferremia develops within hours of infection as part of the acute-phase response. Low serum iron concentrations in the host are thought to limit the proliferation of invading pathogens. A number of acute-phase proteins work to sequester iron in tissues, particularly the macrophages of the spleen. Other responses appear to protect the flow of diferric Tf to erythropoiesis as Cp levels increase, possibly facilitating more efficient transfer of iron to Tf as Tf protein levels in serum decrease, favoring the formation of differic as opposed to monoferric Tf.11
Hepcidin antimicrobial peptide (Hepc) has gained considerable attention for its role in the hypoferremia of AI. Hepc is a peptide hormone that is produced predominantly by hepatocytes, but binds its receptor, Fpn, at distal sites such as duodenal enterocytes and tissue macrophages (Figure 2). Hepc is a potent regulator of iron balance that induces the internalization and degradation of Fpn. Thus, Hepc binding to Fpn on the cell surface restricts dietary iron absorption through the enterocyte and restricts the release of iron recycled by macrophages from senescent erythrocytes.

Circulating hepcidin concentration is regulated by molecular mediators that communicate the status of iron stores (Tf, TfR, TfR2, HFE, HJV, BMP-6, Smad4), inflammation (IL-6), and erythroid drive (GDF15, TWSG, and likely others). Hepcidin promotes the internalization and degradation of Fpn on tissue macrophages. Erythrophagocytosis and inflammation also transcriptionally and translationally regulate Fpn within the macrophage.
Circulating hepcidin concentration is regulated by molecular mediators that communicate the status of iron stores (Tf, TfR, TfR2, HFE, HJV, BMP-6, Smad4), inflammation (IL-6), and erythroid drive (GDF15, TWSG, and likely others). Hepcidin promotes the internalization and degradation of Fpn on tissue macrophages. Erythrophagocytosis and inflammation also transcriptionally and translationally regulate Fpn within the macrophage.
Consistent with its role as a negative regulator of iron absorption, Hepc is induced in response to increasing iron stores. The proteins encoded by the hereditary hemochromatosis genes HFE and TfR2 are modified by their interactions with Tf and TfR to modulate hepcidin production from hepatocytes.12 Likewise, hemojuvelin (HJV) regulators, which include bone morphogenic protein 6 (Bmp6), 13 neogenin,14 and the intracellular signaling molecule Smad4,15 are also required for appropriate hepcidin expression. Hepcidin expression is suppressed in response to increased erythroid drive.16 Growth differentiation factor 15 (GDF15) and twisted gastrulation 1 (Twsg1)17 are two transforming growth factor-beta (TGFβ) family products generated during ineffective erythropoiesis that also down-regulate hepcidin. While their expression may be limited to patients with ineffective erythropoiesis, they provide support for a new paradigm by which erythroid-precursor derived products affect hepcidin expression.18 Soluble HJV19 and transmembrane protease, serine 6 (TMPRSS6)20,21 have also been shown to negatively regulate hepcidin expression. TMPRSS6 can cleave HJV, but whether the soluble form of HJV or the cleaved membrane-associated portion of HJV transduce the signal to the hepcidin promoter is not yet clear. In addition to these competing systemic signals for Hepc regulation and post-translational control of Fpn by hepcidin, Fpn mRNA levels can be increased by erythrophagocytosis22,23 or decreased by inflammation at the individual macrophage level.24,25 Thus, plasma iron levels are influenced by Hepc-induced endocytosis of Fpn, as well as by potentially competing signals driving Fpn synthesis (Figure 2).
Erythroid Precursors
The production of inflammatory cytokines varies between acute and chronic disease states, and can be characterized by phases of pro-inflammatory cytokine production and anti-inflammatory cytokine production. The contribution of individual cytokines can also vary depending on the pathologic processes in each disease. In light of this complex environment, a number of studies have sought to determine which cytokines are most closely linked to the anemia associated with inflammation. Separate studies focused on patients with systemic lupus erythematosus26 and rheumatoid arthritis27 identified the type II acute-phase-response cytokine, interleukin-6 (IL-6), as the cytokine that correlates most closely with hemoglobin concentration, but molecular mechanisms driving this interaction were not investigated. Other cytokines, such as tumor necrosis factor-alpha (TNFα) and IL-1, have been implicated in the inhibition of multiple stages of erythroid development.2
Recently, an alternative splice-variant of Fpn that does not contain an iron-responsive element was identified.28 Fpn1B expression is uniquely enriched in erythroid precursors, although its role is not well understood.29 It has been speculated that baseline inefficiency of iron utilization due to increased uptake with simultaneous efflux through ferroportin may provide a buffer to protect the developing erythrocyte from iron deficiency in inflammation, where increased hepcidin blocks iron efflux from erythrocytes. However, hepcidin has been shown to negatively regulate the development of erythroid precursors in vitro,30 raising the possibility of more complex effects.
Erythrocyte Survival
Intriguing evidence for increased erythrocyte turnover in animal models of chronic inflammation31 and chronic disease is available,32 but erythrocyte survival has been studied in only a few patient groups because of the technical difficulties of such studies. A newly developed, noninvasive method to measure carbon monoxide (derived from degraded heme) in the breath may improve our ability to assess erythrocyte survival in patients with chronic disease.33
Tools for Diagnosis
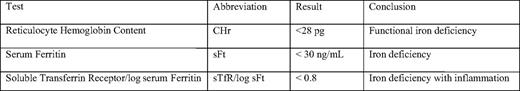
Sensitive and specific clinical assays are essential for the diagnosis of AI and for directing a physician toward the optimal treatment course. The World Health Organization defines anemia as Hgb < 13 g/dL in men and Hgb < 12 g/dL in women. Thus standard parameters from the complete blood count with differential, such as hemoglobin, are essential. Generally, mean cell volume (MCV) and mean cell hemoglobin (MCH) are normal, but can drop as the disease persists. Reticulocyte hemoglobin content (CHr) is likely to become more widely available than it is currently. CHr is a measure of hemoglobin in the most recently produced erythrocytes. Due to the long life span of erythrocytes, processes that restrict the synthesis of Hgb may manifest as decreased CHr much before the effect is seen in the general population of erythrocytes. The measurement of CHr is a very sensitive diagnostic tool that is useful in the early detection of iron-deficiency anemia, and it may also prove useful when monitoring patients with acute infections or chronic diseases who are at risk for AI. CHr < 28 pg (Table 1) is consistent with functional iron deficiency.34 Red cell distribution width (RDW) indicates erythrocyte heterogeneity. A recent study suggests that increased RDW in heart failure is related to inflammatory stress and impaired iron mobilization,35 indicating that it might also be useful in the diagnosis of AI, although this particular diagnostic has not been thoroughly studied in this context.
The presence of inflammation is often inferred by the presentation of a given infection or disease state, but elevated neutrophils, monocytes, and platelets also indicate inflammation. The detection of C-reactive protein has improved with the availability of high-sensitivity enzyme-linked immunosorbent assays (ELISAs), but remains a fairly nonspecific measure of inflammation. Many ELISAs are available for analysis of individual pro-inflammatory cytokines. Either individually or in multiplex form, these ELISAs may eventually help to discriminate between the mechanisms that drive AI.
Serum iron and Tf saturation are decreased in AI, indicating that the iron supply to the erythron is limited. Confirmation of sufficient iron stores can be difficult. When a bone marrow aspirate is available, macrophage iron stores can be confirmed by Prussian blue iron staining, but less invasive diagnostics have become the standard of care. Serum ferritin (sFt) is generally considered a marker of iron stores. Mounting evidence suggests that it is primarily produced by macrophages,36 but other cell types may contribute to sFt. Generally, when sFt is below 30 ng/mL in an anemic patient, iron deficiency can be diagnosed.2 However, in many patients, a combination of iron deficiency and AI may exist. sFt is induced in response to inflammation. Thus, in the context of AI, it is a poor marker of available iron. sFt has been used in conjunction with the serum transferrin receptor (sTfR) to determine the sTfr/log sFt ratio. sTfR is produced when erythroid precursors have produced sufficient hemoglobin and shed the receptor. As iron availability to the erythron decreases, or as erythroid capacity increases, sTfR in the plasma increases. Despite iron restriction in the context of AI, sTfR is not elevated, but remains in the normal range due to down-regulation by pro-inflammatory cytokines.37 The sTfR/log sFt ratio can discriminate between iron-deficiency anemia and AI. sTfR/log sFt > 1.5 is most consistent with iron deficiency, but sTfR/log sFt < 0.8 best defines iron deficiency with inflammation34 (Table 1). This diagnostic determination is currently limited in availability and is underutilized in clinical practice.
Recently, several hepcidin assays have become available.38 A combination of surface-enhanced laser desorption/ionization time-of-flight and mass spectroscopy (SELDI-TOF-MS) has been used to assess serum and urine Hepc levels in a number of disease states. A competitive ELISA has also been developed for quantitative assessment of Hepc from serum and urine.39 Hepc tends to be correlated most closely with sFt, reflecting its regulation by both inflammation and iron stores. While an ELISA for the 83-amino-acid pro-hepcidin is available, this form of Hepc is not bioactive and does not correlate with the 25-amino-acid, bioactive form of Hepc in the serum.
Currently, there are no established standards or cutoffs for Hepc-based diagnoses.38 Hepc levels must be interpreted in the context of the physiology of the individual. One might argue that an anemic adult with Hepc concentrations in the normal range has inappropriately elevated Hepc, because it should be low in response to hypoxia. Several investigators have suggested methods to normalize Hepc values based on other biomarkers such as sTfR, serum Fe, or sFt. However, because reliable methods for measures of Hepc have only recently become available, more work must be done to assess Hepc levels in various disease states before appropriate diagnostic guidelines can be developed.
Treatment
The primary goal for the treatment of AI is to treat and cure the underlying source of inflammation. However, when the inflammatory insult is chronic, poorly controlled, or independent of any identified disease state (such as in the context of aging), alternative treatment strategies are valuable.
Recently, the Dialysis Patients' Response to IV iron and with Elevated Ferritin (DRIVE) trial has tested the hypothesis that functional iron deficiency due to inflammation drives Epo resistance in dialysis patients.40 Subjects were carefully selected to avoid active infections, and then those with transferrin saturation under 25% and sFt of 500 to 1200 ng/mL were randomized to receive or not receive intravenous iron. Both groups received a 25% increase in erythropoiesis-stimulating agent. Ferric gluconate improved hemoglobin production in this patient population compared with controls.
Despite baseline elevated sFt, hemoglobin concentration increased in DRIVE subjects receiving additional intravenous iron. In light of this result, other studies aimed at increasing iron availability for patients with anemia due to other chronic disease states should be considered. These data also suggest that small molecules that inhibit hepcidin activity or Fpn internalization and increase systemic iron may also be beneficial. Such an approach would also be superior to ferric gluconate, because it would reduce the risk of oxidative stress resulting from additional iron.
Future Directions
Our understanding of the role of hepcidin in AI is rapidly growing. In order to benefit fully from the expanding knowledge base, a method for standardizing hepcidin measurements is essential. While hepcidin plays a central role in systemic iron metabolism, other factors may contribute to AI. More thorough translation of the CHr and sTfR/sFt indices to clinical practice could potentially improve the detection of AI in time for more flexible treatment options for patients. New methods for noninvasive analysis of erythroid maturation are still needed. The identification of GDF15 as a novel biomarker for ineffective erythropoiesis is a strong indication that similar markers for normal erythropoiesis must exist. Novel assays, such as the carbon-monoxide breath monitor for the assessment of erythrocyte survival, are appealing and should be applied more broadly across various disease states.
As we continue to develop tools for timely diagnosis and targeted treatment of AI, we must keep in mind that AI is a highly developed part of the innate immune response. Anemia tends to correlate with poor outcomes across a number of disease states, but this most likely reflects the severity of the disease. As the field moves forward, it will be important to understand whether treatment of AI improves disease outcomes for subjects in the long run.
Acknowledgments
This work was supported in part by NIH grants DK 082722, AG021334, and AG029148, and by the American Society of Hematology Scholar Award.
Disclosures
Conflict of interest disclosure: none. Off-label drug use: Ferric gluconate is not approved for the treatment of anemia associated with inflammation
Correspondence
Cindy N. Roy, PhD, American Society for Hematology Scholar, Assistant Professor of Medicine, Division of Geriatric Medicine and Gerontology, Johns Hopkins University School of Medicine, 5501 Hopkins Bayview Circle, Room 2A.44, Baltimore, MD 21224; Phone: (410) 550-9941; Fax: (410) 550-2513; e-mail: croy6@jhmi.edu
