
Abstract
Autoimmune thrombocytopenia (ITP) is characterized by autoantibody-mediated platelet destruction that can be demonstrated by shortened radiolabeled platelet survival. An additional role of ineffective thrombopoiesis was suggested by autologous platelet kinetic studies performed in the 1980s. Sera of patients with ITP have been demonstrated to inhibit megakaryocyte growth in culture supporting the concept of suboptimal platelet production as a contributing factor to the thrombocytopenia. The relatively modest rise in thrombopoietin (TPO) levels in thrombocytopenic patients with ITP has helped to identify the TPO receptor as a potential target for the treatment of ITP. Initial studies with recombinant TPO in patients with ITP were encouraging, and novel compounds designed to stimulate the TPO receptor and resultant pathways have been shown in randomized trials to be effective in raising the platelet count and sustaining it at safe levels. Adverse effects of these agents have been relatively mild, although rare serious events including increased bone marrow reticulin deposition, increased numbers of circulating blasts and thrombosis have occurred, and theoretic risks of stimulation of megakaryocytopoiesis and platelet activation remain a concern. As these agents become available it will be important to identify those patients who will most benefit from their use. The place of these drugs in the current management algorithms of ITP will evolve over time as results of clinical trials with these agents and experience with their use in the clinic clarify short-term and long-term efficacy and potential toxicities.
Introduction
“Idiopathic” thrombocytopenic purpura described the phenomenon of decreased numbers of platelets in an otherwise healthy person with an apparently morphologically normal bone marrow with plentiful megakaryocytes. In 1951 Harrington reported on the occurrence, sometimes within hours, of a rapid fall in the platelet counts of normal volunteers when injected with blood from 8 of 10 patients with the disorder.1 The discovery that it was the globulin fraction of the blood that contained the “thrombocytopenic factor” and later identification of that factor as an “anti-platelet antibody” was consistent with the model of pathologic destruction of platelets by a self-reactive immune system and a marrow that was unable to sufficiently increase production to compensate. This model of autoimmune thrombocytopenic purpura (ITP) has gone largely undisputed and has shaped our understanding of the disorder and our approach to therapy. Recent studies paint a broader picture of immune dysregulation leading not only to accelerated platelet destruction, but to abnormalities in mega-karyocyte growth and development and poorly compensated thrombopoiesis. These findings open new avenues of possibilities in the management of thrombocytopenia in ITP.
Platelet Kinetic Studies
Dameshek and Miller made the first attempt to measure platelet production in 19462 when they scored the morphologic appearance of megakaryocytes and platelet budding from patients with chronic and acute ITP, liver disease and splenomegaly. They noted that megakaryocytes were present in increased numbers in the bone marrow of patients with ITP, but were frequently immature appearing and showed a “greatly diminished productivity of platelets,” postulating an abnormality of the spleen exerted “an unusual effect upon the production of platelets from the megakaryocytes.”
A more dynamic measurement of platelet production requires an understanding of the “steady state,” i.e., when the peripheral blood platelet count is stable. At a stable count, platelet production must equal platelet destruction and the number of platelets going into or out of the system every day, “turnover,” must be constant. Harker and Finch first described methodology for 51Cr labeling platelets to measure platelet disappearance rates and, by implication, production.3 The number of platelets entering and leaving the circulation every day can be calculated by the formula:
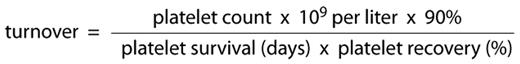
At a given platelet count, the shorter the platelet survival, the smaller the denominator and the higher the resultant turnover (production rate). Harker and Finch performed platelet survivals in 15 normal subjects and 4 patients with ITP (Table 1 ). Survivals of radiolabeled donor platelets were markedly shortened in the patients with ITP, from a mean of 9.9 days in normal subjects to 48 to 230 minutes. Calculated turnover rates in patients with ITP were 4 to 9 times normal. Platelet kinetics in 7 patients with secondary autoimmune thrombocytopenia were similar to the ITP group. Branehog later confirmed these findings.4
Improved labeling techniques allowed the use of autologous platelets in later experiments,5,6 avoiding potential confounding and adverse effects of allogeneic platelets. In dual-labeling experiments, Ballem and colleagues compared 111In- and 51Cr-labeled autologous and homologous platelet survivals in 13 patients with chronic ITP and found autologous platelets survive significantly longer in the circulation than do homologous platelets. Longer platelet survival implies lower destruction and turnover rates, suggesting an inability to effectively increase platelet production. In these experiments, platelet production by the megakaryocyte was not measured directly, and an alternative explanation is that circulating autologous platelets in patients with ITP are relatively protected, the majority of destruction intramedullary or occurring shortly after release from the marrow.
Thrombopoietin and ITP
In 1994 TPO was first characterized by several teams of investigators.7–10 TPO binds to its receptor (c-Mpl) on the megakaryocyte membrane and stimulates platelet production and megakaryocyte proliferation and differentiation via phosphorylation of JAK2 and STAT5, and MAPK. TPO is primarily produced by the liver and one suggested model of TPO regulation is a simple feedback loop by which TPO is bound onto high-affinity receptors on the platelet membrane and internalized.11,12 At high platelet counts, TPO is largely taken up by the platelets and little is left circulating free to bind to megakaryocyte TPO receptors and stimulate proliferation and platelet production. A fall in platelet count results in increased TPO and increased megakaryocyte c-Mpl binding. As expected, levels were found to be increased in amegakaryocytic thrombocytopenias, but surprisingly minimally if at all elevated in patients with chronic ITP.13,14 A study of more than 200 ITP patients found evidence of anti-TPO antibodies in only 1 patient,15 although antibodies against the TPO receptor have been detected in ITP patients and thrombocytopenic patients with systemic lupus erythematosus.16
Cell culture experiments
Glycoprotein surface antigens co-expressed on platelets, megakaryocytes and megakaryocyte precursors (gpIIb-IIIa and gpIb-IX) are recognized by autoantibodies from ITP patients,17 and cell culture experiments utilizing plasma from ITP patients demonstrate inhibition of megakaryocyte growth and development. Nugent’s group found umbilical cord blood mononuclear cells yield fewer mega-karyocytic cells if incubated with plasma from ITP patients containing anti-glycoprotein Ib, IIb or IIIa,18 and McMillan reported that plasma from 10 of 18 ITP patients added to cell cultures resulted in a significant decrease in total numbers of megakaryocytes and maturation inhibition with decreased ploidy when compared to control cultures.19 Adsorption of platelet autoantibody by immobilized antigen attenuated the inhibition.
Li and coworkers cultured bone marrow mononuclear cells from patients with chronic ITP and found megakaryocyte counts increased when in the presence of autologous CD8+ T cells while platelet production was reduced compared to controls.20 Lower percentages of polyploidy and apoptotic megakaryocytes were also observed. Dexamethasone added to the coculture corrected the effect of autologous CD8+ T cells on megakaryocytopoiesis. This latter finding is of particular interest considering that the increase in platelet count seen with corticosteroid therapy was not accompanied by an increase in radiolabeled autologous platelet survival,21 suggesting the count rises by increased release of platelets into the circulation in response to corticosteroids.
Thrombopoietic Growth Factors: A New Approach to Raising the Platelet Count in ITP
Traditionally, ITP therapy has targeted the immunologically mediated destruction of platelets with the use of immunosuppressive agents (e.g., corticosteroids, azathioprine) immunomodulation (e.g., gamma globulin preparations, Staph Protein A columns, monoclonal antibodies), and removal of the site of destruction (splenectomy, splenic irradiation). Findings of a curtailed increase in thrombopoietin and impaired megakaryocyte maturation and platelet production suggested the possibility of megakaryocyte stimulation as a treatment strategy for ITP.
A pilot study with recombinant interleukin (IL)-11, known to stimulate megakaryocyte proliferation in vitro, was unsuccessful in raising the platelet count in any of 7 patients with severe chronic ITP.22 Recombinant human megakaryocyte growth and development factor (rHu-MGDF) composed of the amino-terminal 163 amino acids of human TPO had a circulating half-life too short for clinical usefulness until coupled to a polyethylene glycol moiety (PEG-rHuMGDF). When PEG-rHuMGDF was administered daily for 7 days, 3 of 4 patients with ITP experienced a significant rise in their platelet counts.23 An additional patient with cyclic immune thrombocytopenia responded to twice weekly treatment with PEG-rHuMGDF for more than 6 years.24 Unfortunately trials had to be stopped when 13 of 538 normal volunteers and 4 of 650 oncology patients treated with the drug on more than one occasion developed thrombocytopenia.25 Three subjects were found to have antibody against PEG-rHuMGDF that crossreacted with native TPO.26 Although sequence homology with TPO limited the clinical usefulness of this molecule, the principle had now been founded.
A number of candidate molecules were identified by screening of peptide libraries for sequences that bind the TPO receptor and stimulate growth of TPO-dependent cell lines. Several agents have shown promise for clinical purposes in humans and a few have advanced to clinical trials (Table 2 ). Patients admitted to the ITP trials described below have generally had ITP for at least 6 months and had baseline platelet counts < 30 × 109/L at the time of study entry. All had been treated with at least one other modality and were unable to sustain a higher platelet count without some form of therapy and many required ongoing therapy. To date all reported clinical trials have focused only in adults although trials in children are ongoing.
TPO peptide mimetics
Romiplostim (AMG531) is a rationally designed novel molecule, a “peptibody” composed of 2 identical peptide sequences linked via polyglycine and covalently bound with 2 Fc fragments to prolong its circulating half life.27 It has a high affinity for the TPO receptor, induces phosphorylation of JAK2 and STAT5 in TPO-dependent cell lines and increases megakaryocyte differentiation. It is administered as a subcutaneous injection weekly and increases platelet counts in healthy volunteers with a dose-dependent rise in platelet count starting at day 5 and peaking at days 12 to 15. In a Phase I/II study 7 of 12 ITP subjects with platelet counts < 30 × 109/L (or <50 × 109/L if on a stable dose of corticosteroids) had at least a doubling of their platelets to above 50 × 109/L after 2 doses ≥ 3 μ g/kg and 10 of 16 subjects reached a target range of 50 to 450 × 109/L at a stable dose of 1 or 3 μ g/kg for 6 weeks.28
In two parallel placebo controlled randomized trials, 63 splenectomized and 62 nonsplenectomized ITP patients with platelet counts < 30 × 109/L (mean 16 × 109/L) were treated with weekly subcutaneous injections of romiplostim or placebo for 24 weeks.29 A target platelet count of 50 × 109/L was achieved by 25% of patients after 1 week and by 50% within 2 to 3 weeks of beginning romiplostim therapy. Forty-nine percent of romiplostim-treated patients and 2.4% of placebo-treated patients achieved a durable platelet response, defined as a platelet count ≥ 50 × 109/L for more than 6 of the last 8 weeks of treatment. Eighty-three percent of romiplostim treated patients achieved a platelet count of ≥ 50 × 109/L for at least 4 weeks of the study, while 7% of the placebo-treated patients achieved this endpoint. There was no relationship between response and baseline TPO levels. Thirty-one percent of patients were receiving concurrent therapy for their ITP at baseline (37% placebo, 29% romiplostim). Thirty-five percent of romiplostim-treated patients were able to reduce the doses of those medications by ≥ 25% and 52% discontinued them completely. Nineteen percent of subjects on placebo achieved either of those endpoints. Rescue therapy to increase the platelet count to treat or prevent bleeding was required by 59.5% of placebo subjects and 21.7% of patients on the romiplostim arm during the 24-week period. The overall incidence of any bleeding does not appear to be lower in ITP subjects treated with romiplostim but moderate (8%) and severe, life threatening or fatal (8%) bleeding was less frequent than in subjects receiving placebo (24% and 10%).30 All moderate bleeding occurred at platelet counts < 50 × 109/L and severe or worse bleeding at counts < 20 × 109/L. The two fatal bleeding events, one in each arm, occurred at platelet counts of <10 × 109/L.
The majority of reported adverse events with romiplostim treatment have been mild to moderate and have not differed from placebo, but serious adverse events have been reported.30 Increased bone marrow reticulin has been reported in 8 patients who have received romiplostim. Of 5 patients who have had follow-up bone marrows evaluated, partial resolution was noted in 2 and no significant changes were noted in 3 patients. Thrombotic events have been reported in study subjects with equal frequency in both placebo (4.3%) and romiplostim (4.4%) arms. Patients who experienced thrombotic events were not distinguished by more marked increases in their platelet count than those without these events, and all had preexisting risk factors for thrombosis and/or a history of a thromboembolic event. No subject has developed neutralizing antibodies to TPO although anti-TPO antibodies were reported in a small number of subjects before and after treatment.30
Romiplostim continues to be administered to a large number of ITP subjects in an extended open-label trial for continued surveillance of efficacy and safety as well as trials for thrombocytopenia in myelodysplastic syndrome and chemotherapy-induced thrombocytopenia31 and has been approved by the FDA for treatment of chronic ITP.
TPO nonpeptide mimetics
Eltrombopag (Promacta, SB497115) is an extremely small (MW = 442 D) orally available nonpeptide molecule that has been shown to stimulate TPO-dependent cell lines via JAK2- and STAT-signaling pathways to proliferation, mega-karyocyte differentiation and platelet production.32 It interacts with the transmembrane portion of the TPO receptor rather than at the site of native TPO binding. Its absorption can be significantly affected by food and needs to be taken 2 hours before or after meals, and because of its propensity to bind to divalent cations must be taken 6 hours from antacids, calcium or vitamin supplements. When administered to healthy male volunteers daily for 10 days a dose-dependent increase in platelet count began after 8 days and peaked at 16 days.33
In a 6 week placebo-controlled trial evaluating eltrombopag in 118 adults with chronic ITP and platelet counts < 30 × 109/L34 a significantly higher proportion of patients receiving 50 mg or 75 mg of eltrombopag achieved a platelet count of ≥ 50 × 109/L (70% and 81%) compared with those receiving placebo (11%). More than 80% of the eltrombopag-treated patients had reached this endpoint by day 15. In an ongoing open label extension trial with 108 patients 80% achieved a platelet count of ≥ 50 × 109/L at least once, 54% for ≥ 10 weeks and 24% for ≥ 25 weeks.35 Of 40 patients on concomitant ITP medications at study entry, 14 were able to stop therapy. The incidence of bleeding in the placebo group was 14%, 7% and 4% in the 50 mg and 75 mg eltrombopag groups. Nineteen percent of patients reached a platelet count of > 400 × 109/L.
Safety data from 4 randomized, double-blind, placebo-controlled Phase II or III studies of eltrombopag that included 485 thrombocytopenic patients with ITP (n = 231), hepatitis C virus (HCV) infection or chemotherapy-induced thrombocytopenia has been reported.36 Most adverse events were mild and did not differ from the placebo group. Cataract development occurred in 7 patients (1 placebo) and cataract progression in 5. Of the 6 patients with cataracts who had been treated with eltrombopag for ITP, all had been treated in the past with corticosteroids for some length of time. Of 231 patients with ITP treated with eltrombopag (n = 170) or placebo (n = 67) for 6 weeks in Phase II and III trials, 1 (eltrombopag arm) experienced a pulmonary embolism and thromboemboli. The long-term safety of eltrombopag in ITP is being evaluated in an ongoing, open-label extension trial (EXTEND). Data have been reported on 109 patients, 30 of them on eltrombopag for at least 24 weeks. Most reported adverse events (n = 109) have been mild in severity, headache being the most frequently observed adverse event (17%). Eight patients developed an increased ALT and hyperbilirubinemia and 2 had pulmonary emboli. Patients with a prior history of arterial or venous thrombosis and known risk factors for thrombosis and patients with a history of cardiovascular disease were excluded from the eltrombopag trials. The presence of reticulin/collagen was reported in 9 of 19 bone marrow biopsies performed on patients on eltrombopag for at least 13 months. Baseline bone marrow evaluations were not available so an association could not be determined.
Eltrombopag has also been evaluated for treatment of thrombocytopenia in patients with HCV infection and has been shown to be effective in raising the platelet count to enable antiviral therapy.37 HCV infection is associated with antibody-mediated platelet destruction and the thrombocytopenia may predate the hepatic damage that can decrease TPO production and testing for HCV may be advisable in the patient with ITP, even if risk factors are not present. Application for short-term use of eltrombopag for patients with chronic ITP is under review by the FDA.38 Trials with eltrombopag in ITP, HCV and chemotherapy-induced thrombocytopenia are ongoing.39
AKR-501 (YM 477) is another nonpeptide agonist that stimulates growth of TPO-dependent cell lines and is specific for only the human and chimp TPO receptors.40 It is orally active with a serum half-life of approximately 16 hours. It has been shown to increase platelet counts in healthy volunteers after single and multiple doses without significant adverse effects.41 Clinical studies with AKR-501 for chronic ITP are ongoing.39
TPO agonist antibodies
Stimulating Platelet Production: Potential Pitfalls
Potential adverse consequences of therapy with thrombopoietic growth factors are listed in Table 3. Although thrombocytosis in the otherwise healthy patient with iron deficiency or post-traumatic splenectomy is generally not felt to be associated with a marked increased risk of thrombosis, sudden increases in the platelet count in ITP patients may be more problematic.44 Prospective screening of 186 adult patients with chronic ITP revealed a history of 18 thrombotic or ischemic events in 10 patients (5%), half of whom had previously undergone splenectomy.15 Thirteen of the events were venous suggesting the possibility of an inflammatory component and 11 occurred after the diagnosis of ITP was made. In-vivo platelet activation, circulating platelet-leukocyte-monocyte aggregates, young “hyper-reactive” platelets and platelet-derived micro-particles have all been implicated in the mechanism of a prothrombotic state in patients with ITP.45,46
Supraphysiologic doses of TPO facilitate platelet adhesion47 and aggregation induced by shear stress and various agonists.48 In contrast, treatment of patients with advanced cancer with recombinant Peg-HuMGDF stimulated production of platelets without producing any evidence of in vivo activation49 and Harker was unable to show a disproportionate increase in 111In platelet or 125I-fibrin deposition50 on endarterectomized aorta and vascular grafts after treatment of nonhuman primates with rHu-MGDF. Romiplostim did not increase platelet aggregability in 24 healthy individuals who received doses of the drug shown to be clinically active in many patients with ITP.51
The rates of thrombotic events in patients with ITP treated with platelet growth factors do not appear to be different from those seen in placebo groups. Although no relationship has been observed between elevated platelet counts and thrombosis in these studies, caution is advised when considering the use of thrombopoietic agents in patients with known risk factors or a history of thrombosis. It is possible that thrombocytopenia is protective in a state where platelets are activated and microparticles circulate and restoring hemostatic equilibrium is a more appropriate goal than a normal platelet count.
The observation of a close inverse relationship between platelet count and bleeding severity requires caution be used when tapering or discontinuing concurrent ITP medications during thrombopoietic growth factor therapy. An increase in the platelet count may be due to the combined effects of diminished platelet destruction by immunomodulatory therapy and increased platelet production by the thrombopoietic agent. Therefore, any decrease in concurrent ITP medications such as corticosteroids should be approached carefully, as a dose increase of the thrombopoietic agent may be required to regain a balance that sustains the platelet count. Since it may take a week or more before the full effect of a change in dose of these agents is observed, subsequent changes in concurrent medications should wait until the count has once again stabilized. Platelet counts can be expected to fall back to baseline after withdrawal of thrombopoietic agents and theoretically could fall lower if endogenous TPO levels are suppressed. When discontinuing thrombopoietic therapy careful monitoring is required with readiness of rescue therapy for severe thrombocytopenia or bleeding if necessary.
Reticulin deposition appears to be a dose-dependent adverse effect of romiplostim therapy related to megakaryopoietic stimulation and not an idiosyncratic treatment complication. Subjects with leukemia treated with recombinant human TPO and GM-CSF exhibited increased marrow reticulin, which resolved fully a mean of 30 days after discontinuation of treatment.52 Plasma levels of transforming growth factor-β (TGF-β) are known to increase following treatment of mice with a recombinant TPO, and other growth factors and cytokines associated with megakaryocytes and platelets may be important mediators of bone marrow reticulin formation. It is likely that with continued therapy increased reticulin deposition is a side-effect of all thrombopoietic agents.
Reports of increased rates of tumor progression with the use of erythroid-stimulating agents for chemotherapy-induced anemia53,54 have roused concerns that hematopoietic growth factor therapy could stimulate tumor cell proliferation. Many hematopoietic malignancies contain the TPO receptor,55 but administration of Peg-rHuMGDF did not result in increased numbers of blasts or affect treatment response in patients undergoing therapy for acute leukemia.56 An increase in neoplasms has not been reported in patients treated with thrombopoietic agents for ITP, but in a trial of romiplostim in 44 patients with low or intermediate-1 risk myelodysplasia, 6 subjects had transient increases in circulating blasts that cleared after the drug was stopped and 1 subject progressed to AML.57
Balancing Platelet Production and Destruction in ITP: A New Paradigm
Emerging therapies targeting platelet production present new opportunities for the management of ITP. These agents have proven to be effective and relatively safe in raising the platelet count for short periods in patients with chronic ITP. However, they do not increase the platelet count as quickly as steroids and immunoglobulin preparations and should not be used alone to acutely raise the platelet count in patients who are bleeding or severely thrombocytopenic. They will be useful in raising the platelet count in a predictable manner for a planned procedure or when patients unresponsive to or intolerant of other agents require temporary increases in their count.
There are only rare reports of sustained increases in platelet counts after discontinuation of thrombopoietic agents58 and they should not be considered interchangeable with treatments that are intended to induce long-term remission or cure. Patients with chronic ITP requiring ongoing therapy should be offered a curative therapy if not otherwise contraindicated. Thrombopoietic agents may be useful in sustaining the platelet count until longer term therapy can take effect. Experience with these agents has necessarily focused on patients with the most severe and refractory disease and there is only limited experience with prolonged exposure to these drugs. For patients who are refractory to “curative therapy,” the decision to treat a patient with a thrombopoietic agent long term to sustain the platelet count above baseline must take into consideration that severe bleeding does not generally occur above platelet counts of > 20 × 109/L and patients with moderate thrombocytopenia (> 30 × 109/L) do well overall.59 When treating patients with these agents a safe platelet count, i.e., 50 to 100 × 109/L, may be a more appropriate aim than a normal platelet count. The lowest dose necessary to maintain the count in this safe range should be used, an important goal being reduction of concurrent ITP therapies and their associated risks. Synergism may exist between thrombopoietic agents and those that decrease platelet destruction, and dose adjustments that may affect kinetic balance should be approached cautiously. Patients should not only be carefully selected but also monitored closely for evidence of marrow abnormalities, thrombosis and as yet unrecognized adverse effects.
Platelet growth factors and indications currently under investigation.
| Romiplostim (AMG531) |
| Thrombopoietin (TPO) peptide mimetic |
| Binds at the endogenous TPO-binding site |
| Weekly subcutaneous administration |
| Eltrombopag (Promacta, SB49711) |
| TPO non-peptide mimetic |
| Binds at the intramembrane portion of the TPO receptor |
| Daily oral administration |
| AKR-501 (YM 477) |
| TPO non-peptide mimetic |
| Binding remote from endogenous TPO-binding site |
| Daily oral administration |
| Indications currently under investigation |
| Autoimmune thrombocytopenia |
| Thrombocytopenia associated with chronic liver disease |
| Thrombocytopenia associated with the treatment of hepatitis C virus |
| Thrombocytopenia induced by chemotherapy for malignancy |
| Thrombocytopenia associated with intrinsic marrow abnormalities |
| Thrombocytopenia associated with the treatment of hematologic malignancy |
| Romiplostim (AMG531) |
| Thrombopoietin (TPO) peptide mimetic |
| Binds at the endogenous TPO-binding site |
| Weekly subcutaneous administration |
| Eltrombopag (Promacta, SB49711) |
| TPO non-peptide mimetic |
| Binds at the intramembrane portion of the TPO receptor |
| Daily oral administration |
| AKR-501 (YM 477) |
| TPO non-peptide mimetic |
| Binding remote from endogenous TPO-binding site |
| Daily oral administration |
| Indications currently under investigation |
| Autoimmune thrombocytopenia |
| Thrombocytopenia associated with chronic liver disease |
| Thrombocytopenia associated with the treatment of hepatitis C virus |
| Thrombocytopenia induced by chemotherapy for malignancy |
| Thrombocytopenia associated with intrinsic marrow abnormalities |
| Thrombocytopenia associated with the treatment of hematologic malignancy |
Possible adverse effects of thrombopoietic growth factor therapy.
|
|
Disclosures Conflict-of-interest declaration: The author receives research funding from MGI Pharma; is a consultant for, is on an advisory committee for, and receives research funding from Glaxo Smith Kline; is a Consultant for, receives honoraria and research funding from, and is on an advisory committee for Amgen. Off-label drug use discussion: At the current time, AMG 531 (romiplostim), Eltrombopag and AKR501 are all experimental agents. One or two of these may have approval for the use that will be discussed (treatment of chronic ITP) by the time of publication.
