
Abstract
Aplastic anemia may be inherited or acquired. The distinction between these lies not in the age of the patient, but in the clinical and laboratory diagnoses. Adult hematologists must consider adult presentations of the inherited disorders, in order to avoid incorrect management of their patients. Physicians for adult patients must also realize that children with inherited disorders now survive to transition into their care. The major inherited bone marrow failure syndromes associated with development of pancytopenia include Fanconi anemia, dyskeratosis congenita, Shwachman-Diamond syndrome, and amegakaryocytic thrombocytopenia. The ages at presentation are highly variable, but often include individuals of adult age who have previously undiagnosed Fanconi anemia or dyskeratosis congenita. Many of the genes responsible for these disorders have been identified (12 Fanconi anemia genes, 3 dyskeratosis congenita genes, and 1 each for Shwachman-Diamond syndrome and amegakaryocytic thrombocytopenia). A high index of suspicion and specific testing of children or adults with what appears to be acquired aplastic anemia may identify inherited disorders. Correct classification of patients with aplastic anemia of any age is mandatory for their appropriate management.
The evaluation of aplastic anemia in children and adults differs, based on the expectation that a large proportion of children will have an underlying genetic etiology, while the majority of adults will have an acquired, non-genetic disease. However, this boundary is unclear, and cannot be defined solely by age. Patients have been diagnosed with inherited bone marrow failure syndromes (IBMFS) in their 60s or later, and infants have been determined to have acquired aplastic anemia. Medical management differs both according to age, for a given etiology, and according to etiology, depending upon the age. Hematopoietic stem cell transplant (SCT) protocols also depend on both the age and the underlying cause of the marrow failure. In addition, several of the IBMFS are associated with a high risk of leukemia and/or specific solid tumors, while patients with acquired aplastic anemia are also at risk of hematopoietic clonal disease (if treated with immunosuppression) or subsequent solid tumors (if recipients of a stem cell transplant). A comprehensive review of all of the syndromes can be found in recent books.1,2
Inherited Bone Marrow Failure Syndromes
The most obvious clue that a patient with aplastic anemia has an IBMFS lies in the physical examination (Table 1 ), which may reveal features unique to each of the syndromes. These include café au lait spots and radial ray anomalies in Fanconi anemia (FA), dyskeratotic finger and toenails in dyskeratosis congenita (DC), abnormal thumbs in Diamond-Blackfan anemia (DBA), and absent radii with thumbs present in thrombocytopenia absent radii (TAR). However, some of the IBMFS do not have a specific phenotype, and some patients in the disorders usually associated with specific phenotypes may appear normal. Furthermore, the perception that these syndromes are all diagnosed in childhood is simply incorrect; the magnitude of under diagnosis in adults is unknown, however. While at least 25% of children with aplastic anemia may have a genetic origin, the estimate of less than 10% in adults may be too low.1
Additional clues may be found in the family histories, which must include information about hematologic diseases, leukemia, solid tumors, and birth defects or other abnormal physical findings. The inheritance patterns of the IBMFS encompass autosomal recessive, autosomal dominant, and X-linked recessive, as well as sporadic cases, and thus drawing of a pedigree should be part of the history (Table 1 ). Personal medical histories are also informative, since many of the IBMFS (particularly FA and DC) may present with syndrome-specific cancers, myelodysplastic syndrome (MDS) or leukemia prior to development of aplastic anemia. Ethnic background is relevant, since there are founder mutations in FA genes in Ashkenazi Jews, South African Afrikaaners, and Spanish Gypsies, for example, which increase the expected carrier rates from 1/300 to 1/100, and the incidence of homozygotes from 1/360,000 to 1/40,000. Further insights may derive from the pre-transfusion complete blood count, in which insidious rather than rapid development of anemia, macrocytosis, and elevated fetal hemoglobin are consistent with a long-standing marrow failure, which is more often genetic than acquired.
Table 2 lists the usual therapies for each of the IBMFS, and Table 3 summarizes the potential evolution to leukemia and syndrome-specific solid tumors. The neoplastic complications of the IBMFS are beyond the scope of this review, but are provided to remind the hematologist that all of the IBMFS are pre-malignant conditions and that even solid tumors may develop in these patients with “benign” hematologic syndromes.
It must be pointed out that the adult hematologist has two challenges with regard to the inherited bone marrow failure syndromes: 1) Patients who appear to have an acquired disorder may in fact have a genetic one; and 2) Patients with known IBMFS may have reached adulthood and now need to be managed by the adult hematologist. This review will focus on the former; I will suggest that management of the patients in both categories be undertaken with the collaboration of physicians with experience in IBMFS, unrelated to the patient’s age.
Pancytopenias
Fanconi anemia
Fanconi anemia (FA, OMIM 227650) is the most common of the IBMFS.1 Although approximately 75% of the known patients with FA have one or more physical abnormalities (Table 1 ), many patients appear normal or have subtle findings that may be overlooked (such as short stature, café au lait spots, and hypoplastic thenar muscles). Approximately 10% of the reported patients were ≥ 16 years of age at diagnosis, and I am aware of patients with aplastic anemia newly diagnosed as FA in their 40s and 50s. We have shown that the presence of significant birth defects correlates with early onset hematologic disease.3 The corollary of this is that patients with FA who lack birth defects may develop bone marrow failure later in life or may never develop marrow failure. Thus the diagnostic challenge is less for the pediatric hematologist, whose FA patients may have birth defects, or who thinks of FA in any child with aplastic anemia. The challenge is rather for the adult hematologist to consider inherited marrow failure syndromes such as FA, and particularly to do so in the presence of a normal physical examination in an older individual.
FA must be thought of in order to make the diagnosis. The usual test is determination of increased chromosome breakage in mitogen-stimulated peripheral blood lymphocytes after culture in the presence of a DNA-crosslinker such as diepoxybutane or mitomycin C. However, at least 10% of patients with FA have hematopoietic somatic mosaicism, in which there has been a molecular genetic correction in a stem cell, leading to reversion to normal in one allele, which in turn confers a growth advantage to the progeny of that stem cell.4,40 Thus, if FA is strongly suspected, a skin biopsy is required for determination of chromosome breakage in cultured fibroblasts. The diagnostic algorithm for FA is shown in Figure 1 .
FA is an autosomal recessive disease. Eleven of the more than 12 FA genes have been cloned to date. The FA gene products collaborate in a complicated pathway whose ultimate role is the repair of DNA damage. An intermediate step involves the formation of a complex of proteins from FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM4a, which in turn catalyzes the ubiquitination of the downstream protein product of FANCD2, which can then participate in DNA damage response foci that include other gene products such as BRCA1, AT, NBS, Mre11, etc.5,6 The story is further complicated by the identification of FANCD1 as BRCA27 and FANCJ as BRIP1, which interacts with BRCA1.41–44FANCI has been identified but not yet cloned.8 Thus, a research screening test for most FA cases utilizes Western blots in which an antibody to FANCD2 can distinguish the presence or absence of D2 itself, as well as the monoubiquitinated (long) from the initial D2 protein (short).9 In normal individuals with an intact FA pathway, in whom ubiquitin is added to the D2 protein, the Western blot shows 2 bands, while in FA patients there will be only 1 band if the FA pathway defect is before the ubiquitination step.
Several of the FA genes have been cloned into retro-viruses and can be introduced into FA cells in culture. The gene that corrects (complements) baseline chromosome breakage or poor cell growth in the presence of DNA damaging chemicals is thus inferred to be the gene that was mutant in those cells, and determines the patient’s complementation group. Mutations can then be identified in the appropriate gene by sequencing. Given the amount of effort required for the mutation analyses, most patients with aplastic anemia are first screened for FA by chromosome breakage as shown in Figure 1 .
Failure to correctly diagnose FA may be fatal to the patient. To my knowledge, patients with aplastic anemia due to FA do not respond to immunosuppressive therapy, and precious time may be lost during such treatment. The FA consensus guidelines suggest initiation of treatment when the Hb is ≤ 8 g/dL, platelets ≤ 30,000/fL, and neutrophil count ≤ 1000/fL (www.fanconi.org). If there is an HLA-matched sibling, SCT is the recommended course. In the absence of a matched sibling, SCT can be considered if there is a suitable alternative donor, although this is a higher risk procedure than with a matched sibling. SCT protocols in FA are modified from those used in acquired aplastic anemia, because of the sensitivity of the non-hematopoietic tissues in FA to DNA damage. Modifications include using low-dose cyclophosphamide and no irradiation, or fludarabine-based protocols.10,11 Patients with FA have a high risk of acute leukemia and solid tumors, especially head and neck squamous cell carcinoma, and the risk of the latter is increased further following SCT. There is an association with severe graft-versus-host disease, which might be reduced with the less toxic preparative regimens.12
Approximately 50% of patients with bone marrow failure due to FA improve when treated with androgens. These improvements may be only in Hb, or bi- or tri-lineage, and may be brief or last for many years. Severe neutropenia may respond to granulocyte-colony stimulating factor (G-CSF), and some patients also demonstrate an increase in Hb and platelets with this treatment.13 While gene therapy is tantalizing in FA, it is not yet a reality.
Dyskeratosis congenita
Dyskeratosis congenita (DC) is the IBMFS in which the majority of the patients are actually diagnosed as adults, because the diagnostic triad (dyskeratotic nails, lacey reticular rash, and oral leukoplakia) often develops during teen or young adult ages. Here the diagnostic and therapeutic challenge lies with the pediatric hematologist, who may be treating a young child with severe aplastic anemia who does not yet have the physical findings seen in DC. These patients do not respond to immunosuppression, and following an apparently successful SCT may develop findings that resemble chronic graft-versus-host disease but are actually hallmarks of DC. The abnormal nails are often the first sign, but may be misinterpreted as fungal infection in a neutropenic patient. Another early sign is epiphora from lacrimal duct stenosis.
DC appears clinically to be X-linked recessive (OMIM 305000), autosomal dominant (OMIM 127550), or autosomal recessive (OMIM 224230), and the family history may be informative.14,15 The majority of male cases that appear to be X-linked are due to mutations at Xq28 in DKC1, which codes for dyskerin, a protein that is involved in the telomere maintenance pathway. Some, but by no means all, of the patients in autosomal dominant families have mutations at 3q26 in TERC, the RNA template in the telomerase complex. In many DC families the relevant gene appears to be neither DKC1 nor TERC, however, and no mutant genes have been identified for the families that appear to be autosomal recessive. The phenomenon of “anticipation” has been described in DC, and thus signs and symptoms may appear earlier in younger generations, while their relatives may develop problems as older adults.16
Recently, several groups have observed that some patients with apparently acquired aplastic anemia (and failure to respond to immunosuppression) have very short telomeres in their peripheral blood white cells, and some of these were found to have mutations in the TERC gene.17–19 These mutations were also found in some family members who did not have aplastic anemia. Neither the probands nor their family members had the physical features of DC. Further investigations identified other patients with aplastic anemia and no clinical phenotype (who had short telomeres in blood cells) as having mutations in TERT, the gene for the telomerase reverse transcriptase enzyme.20 In the aplastic anemia cohort seen at the NIH, the prevalence of TERC and TERT mutations was 2.5%, and 3.5% respectively. The authors suggested that TERT and TERC mutations are genetic risk factors for aplastic anemia, and may increase susceptibility to environmental insults. In an accompanying editorial, Fibbe suggested that telomere length should be measured in patients with aplastic anemia who fail immunosuppression, and those with family histories of aplastic anemia.21 Furthermore, it would appear that patients with mutations in any of the genes in the telomere maintenance pathway might in fact be labeled “dyskeratosis congenita with variable expression” rather than “familial aplastic anemia.” This suggestion is reinforced by the observation of missense mutations in TERT in a few patients who appeared clinically to have DC.22 The caveat with regard to screening all patients with aplastic anemia for DC, however, is that the telomere length assay is currently only performed in research laboratories, and thus is usually available only for patients in whom DC is already highly suspected.
Management of DC patients is similar to that for FA. The first-line therapy is usually androgens, followed by G-CSF alone or with erythropoietin.23 While SCT is tempting, the results have until recently been quite poor.1 More recently, non-myeloablative transplants have been reported with good short-term outcomes, but we await larger series and longer follow-up.24 Although transplant teams have been reasonably successful in developing modified SCT protocols for patients with FA, they have not yet identified the ideal approach to patients with DC. This strengthens the need to identify DC (or mutations in genes in the telomerase pathway), whether the patient is called “DC” or “familial acquired aplastic anemia with mutations in TERC or TERT,” because the SCT protocols used for acquired aplastic anemia may be toxic in patients with DC.
Shwachman-Diamond syndrome
Patients with Shwachman-Diamond syndrome (SDS, OMIM 260400) usually present in early childhood with malabsorption due to pancreatic insufficiency, and neutropenia.25 However, a substantial proportion go on to develop aplastic anemia, MDS, or leukemia.1 These complications may occur in SDS patients who have reached adult age, and thus may have outgrown the care of a pediatric hematologist.
SDS is an autosomal recessive disorder, in which the majority of the tested patients have been found to have mutations in the Shwachman Bodian Diamond syndrome gene (SBDS) located at 7q11.26 Pancreatic insufficiency can be confirmed by demonstration of low serum trypsinogen in young children, although this may improve with age and be normal in adults with SDS. More specific is a low serum isoamylase, which increases in normal children until age 3 but remains low in older children and adults with SDS.27 The diagnosis of neutropenia requires documentation at least 3 times, but may improve with age. About half of the reported SDS patients had metaphyseal dysostosis, and short stature unrelated to malabsorption is a common component of the syndrome. Approximately 40% of the reported patients with SDS developed additional cytopenias, including aplastic anemia, at up to 35 years of age. SDS patients with neutropenia may respond to G-CSF, while pancytopenia may require androgens and consideration of SCT. Unfortunately, the survival after SCT is around 50%, unrelated to whether the donor is a matched sibling or an alternative donor.1 Deaths were related to complications of MDS or leukemia, as well as to cardiotoxicity from cyclophosphamide.
Availability of mutation testing in the SBDS gene may now facilitate consideration of SDS in adult patients with neutropenia or aplastic anemia who were not diagnosed in childhood, but may have a family history or personal history of symptoms consistent with this diagnosis.
Amegakaryocytic thrombocytopenia
Amegakaryocytic thrombocytopenia (OMIM 604498), abbreviated Amega, or CAMT (for congenital amegakaryocytic thrombocytopenia), is usually diagnosed in early childhood, due to presentation with isolated nonimmune thrombocytopenia, with decreased marrow megakaryocytes.1 These patients often evolve to full-blown aplastic anemia, MDS, or leukemia. It is an autosomal recessive disorder, and biallelic mutations in the thrombopoietin receptor, MPL, at 1p34, provide a definitive diagnosis for almost all patients.28 The German group found that patients with mutations leading to loss of mpl function all went on to develop early pancytopenia, while those with missense mutations had a delayed or milder course. The only cure for this disorder is SCT, with very good short-term survivals reported.29 As survivors of this procedure age, they may require follow-up by adult hematologists for the risk of late graft loss or leukemia. While it seems unlikely that patients with this condition will present first in adulthood, there are no data to demonstrate that mutations in MPL have no role in adult onset aplastic anemia.
Single Cytopenias
Patients with inherited forms of single cytopenias are usually diagnosed in childhood. This section will briefly touch on those who are identified as adults, as well as those in whom development of pancytopenia has been reported.
Diamond-Blackfan anemia
Diamond-Blackfan anemia (DBA, OMIM 105650) is a disorder with pure red cell aplasia, in which 90% of patients are diagnosed in the first year and reports of newly diagnosed older adults are anecdotal.1,30 However, DBA has also been identified in older members of families of affected children. The physical findings of DBA may include short stature and abnormal thumbs (hypoplastic, triphalangeal, or with underdeveloped thenar muscles), but many patients are normal in appearance.1 The anemia is usually macrocytic, with elevated fetal hemoglobin, and increased red cell adenosine deaminase; these features have been observed in non-anemic relatives.31 Mutations in the gene for small ribosomal protein (RPS19), located at 19q13.2, were found in approximately 25% of DBA patients, and we have also seen mutations in asymptomatic relatives. Thus, DBA may be responsible for what at first glance appears to be adult pure red cell aplasia (PRCA). In addition, a small proportion of DBA patients may develop pancytopenia,32 although whether this is due to DBA itself, or to complications from multiple transfusions, remains to be clarified.
The majority of patients with anemia due to DBA improve with corticosteroid treatment. Those who require high doses or fail to respond, receive regular red cell transfusions, and eventually need iron chelation. SCT is often recommended for those patients.33 It should be pointed out that 15%–25% of DBA patients undergo a remission, and may not need treatment again, or may relapse after reaching adulthood.
Thrombocytopenia absent radii
Thrombocytopenia absent radii syndrome (TAR, OMIM 27400) is perhaps the only IBMFS which should be diagnosed exclusively in the neonatal period, when thrombocytopenia is observed in an infant with bilateral absence of the radii. While the thrombocytopenia may be initially severe, it usually improves with age, and subsequent aplastic anemia has not been reported.1 However, leukemia has been reported, and I have seen a 20 year old with hypoplastic but not absent radii, in whom thrombocytopenia was first noted during teenage years (her younger sibling had a mild form of TAR). Thus, even this diagnosis needs to be in the repertoire of the adult hematologist. No gene has been identified yet for this apparently autosomal recessive disorder. Treatment during infancy or later for surgical procedures includes platelet transfusions as needed. SCT has been performed rarely for patients whose severe thrombocytopenia did not improve.34
Severe congenital neutropenia
Severe congenital neutropenia (SCN, OMIM 207700) is defined as early onset severe neutropenia, with absolute neutrophil counts below 200/mm3, and severe infections. More than half the patients have dominant mutations in neutrophil elastase (ELA2, located at 19p13.3), while a few have mutations in GFI-1.35,36 Mutations in ELA2 have also been observed in cyclic neutropenia, a condition often identified in adults. While it seems unlikely that a patient with SCN would escape diagnosis until adulthood, it is mentioned for completeness. SCN patients may develop MDS or leukemia, but aplastic anemia has not been reported. SCN patients often improve when treated with G-CSF. Thus, adult hematologists may see SCN patients for whom G-CSF has removed the risk of early neutropenic sepsis; they remain at risk of leukemia.
Acquired Aplastic Anemia
In pediatric hematology, aplastic anemia is considered to be acquired only after all inherited syndromes have been reasonably considered and eliminated. In adult hematology, aplastic anemia is considered to be acquired, period. In fact, children can develop acquired aplastic anemia, with presumably the same etiologies as in adults. There may have been exposure to toxic agents, such as radiation, chemotherapy, or certain drugs or chemicals. Other associations include pregnancy, seronegative hepatitis, and eosinophilic fasciitis. Most cases of aplastic anemia are idiopathic and may be immune-mediated, due to activation of cytotoxic T-lymphocytes and production of interferon-γ and tumor necrosis factor-α.37–39 One recent study identified patient-specific expanded subsets of T-cell-receptor Vβ-chains, suggesting dominant clonal expansion of inhibitory cells.38 It is not clear whether acquired aplastic anemia in children has immune mediation similar to that of adults, however, since the cited studies primarily included adults.
Treatment of acquired aplastic anemia differs from treatment of inherited marrow failure disorders. Successful SCT depends in part on the choice of preparation, either immunosuppressive or myelosuppressive, and requires knowing the underlying disorder. Medical managements are essentially mutually exclusive. Patients with inherited marrow failure syndromes may respond to androgens, or to G-CSF and/or erythropoietin, while these treatments are not usually effective in acquired aplastic anemia. On the other hand, patients with acquired aplastic anemia often improve with immunosuppressive therapy, such as antithymocyte globulin plus cyclosporine, maneuvers which are ineffective in the inherited conditions.
For further discussion of the diagnosis and management of aplastic anemia in adults and in children the reader is referred to the chapters in this volume by Maciejewski and Risitano (on adults) and by Guinan (on children).
Inherited bone marrow failure syndromes.*
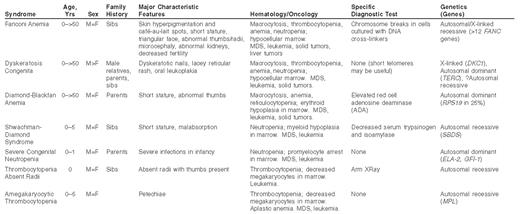
*Some patients with these syndromes have no family history, and none of the physical or hematologic features. MDS, myelodysplastic syndrome.
Management guidelines for inherited bone marrow failure syndromes.
| Syndrome | When to Treat | Pharmaceutical Treatment | Transfusions | Stem Cell Transplant | Spontaneous Improvement |
|---|---|---|---|---|---|
| Abbreviations: Hb, hemoglobin; ANC, absolute neutrophil count; G-CSF, granulocyte colony-stimulating factor. | |||||
| Fanconi Anemia | Hb < 8 g/dL, or ANC < 1000/mm3, or platelets < 30,000/mm3 | Androgens, usually oxymetholone 2–5 mg/kg/day. G-CSF ~5 μg/kg/day. | Packed red cells or platelets as needed | Bone marrow, or cord blood | Rare |
| Dyskeratosis Congenita | Hb < 8 g/dL, or ANC < 1000/mm3, or platelets < 30,000/mm3 | Androgens, usually oxymetholone 2–5 mg/kg/day. G-CSF ~5 μg/kg/day. | Packed red cells or platelets as needed | Bone marrow, or cord blood | Rare |
| Diamond-Blackfan Anemia | Hb < 8 g/dL | Prednisone, 2–5 mg/kg/day | Packed red cells | Bone marrow, or cord blood | ~25% |
| Shwachman- Diamond Syndrome | ANC < 1000/mm3 | G-CSF 5–10 μg/kg/day | Bone marrow, or cord blood | No | |
| Severe Congenital Neutropenia | ANC < 1000/mm3 | G-CSF 5–10 μg/kg/day | Bone marrow, or cord blood | No | |
| Thrombocytopenia Absent Radii | Platelets < 15,000/mm3 | None | Platelets as needed | Bone marrow, or cord blood | Most patients |
| Amegakaryocytic Thrombocytopenia | Hb < 8 g/dL, or ANC < 1000/mm3, or platelets < 30,000/mm3 | Androgens, usually oxymetholone 2–5 mg/kg/day. G-CSF ~5 μg/kg/day | Packed red cells or platelets as needed | Bone marrow, or cord blood | No |
| Syndrome | When to Treat | Pharmaceutical Treatment | Transfusions | Stem Cell Transplant | Spontaneous Improvement |
|---|---|---|---|---|---|
| Abbreviations: Hb, hemoglobin; ANC, absolute neutrophil count; G-CSF, granulocyte colony-stimulating factor. | |||||
| Fanconi Anemia | Hb < 8 g/dL, or ANC < 1000/mm3, or platelets < 30,000/mm3 | Androgens, usually oxymetholone 2–5 mg/kg/day. G-CSF ~5 μg/kg/day. | Packed red cells or platelets as needed | Bone marrow, or cord blood | Rare |
| Dyskeratosis Congenita | Hb < 8 g/dL, or ANC < 1000/mm3, or platelets < 30,000/mm3 | Androgens, usually oxymetholone 2–5 mg/kg/day. G-CSF ~5 μg/kg/day. | Packed red cells or platelets as needed | Bone marrow, or cord blood | Rare |
| Diamond-Blackfan Anemia | Hb < 8 g/dL | Prednisone, 2–5 mg/kg/day | Packed red cells | Bone marrow, or cord blood | ~25% |
| Shwachman- Diamond Syndrome | ANC < 1000/mm3 | G-CSF 5–10 μg/kg/day | Bone marrow, or cord blood | No | |
| Severe Congenital Neutropenia | ANC < 1000/mm3 | G-CSF 5–10 μg/kg/day | Bone marrow, or cord blood | No | |
| Thrombocytopenia Absent Radii | Platelets < 15,000/mm3 | None | Platelets as needed | Bone marrow, or cord blood | Most patients |
| Amegakaryocytic Thrombocytopenia | Hb < 8 g/dL, or ANC < 1000/mm3, or platelets < 30,000/mm3 | Androgens, usually oxymetholone 2–5 mg/kg/day. G-CSF ~5 μg/kg/day | Packed red cells or platelets as needed | Bone marrow, or cord blood | No |
Risk of neoplasia in inherited bone marrow failure syndromes.
| Syndrome | Leukemia | Solid Tumors |
|---|---|---|
| Fanconi Anemia | Acute Myeloid Leukemia | Head and neck squamous cell carcinomas, gynecologic, esophageal, brain tumors |
| Dyskeratosis Congenita | Acute Myeloid Leukemia | Head and neck and anogenital carcinomas |
| Diamond-Blackfan Anemia | Acute Myeloid Leukemia | Osteogenic sarcomas |
| Shwachman-Diamond Syndrome | Acute Myeloid Leukemia | No |
| Severe Congenital Neutropenia | Acute Myeloid Leukemia | No |
| Thrombocytopenia Absent Radii | Acute Myeloid Leukemia | No |
| Amegakaryocytic Thrombocytopenia | Acute Myeloid Leukemia | No |
| Syndrome | Leukemia | Solid Tumors |
|---|---|---|
| Fanconi Anemia | Acute Myeloid Leukemia | Head and neck squamous cell carcinomas, gynecologic, esophageal, brain tumors |
| Dyskeratosis Congenita | Acute Myeloid Leukemia | Head and neck and anogenital carcinomas |
| Diamond-Blackfan Anemia | Acute Myeloid Leukemia | Osteogenic sarcomas |
| Shwachman-Diamond Syndrome | Acute Myeloid Leukemia | No |
| Severe Congenital Neutropenia | Acute Myeloid Leukemia | No |
| Thrombocytopenia Absent Radii | Acute Myeloid Leukemia | No |
| Amegakaryocytic Thrombocytopenia | Acute Myeloid Leukemia | No |

Diagnostic algorithm for Fanconi anemia. FA must be suspected, and the appropriate tests ordered, depending on the level of suspicion.
Diagnostic algorithm for Fanconi anemia. FA must be suspected, and the appropriate tests ordered, depending on the level of suspicion.
