
Abstract
A variety of factor concentrates are currently available for replacement therapy for patients with hemophilia. These differ by several parameters, including source (pooled from pooled blood vs recombinant), purity, pathogen inactivation, and by the presence or absence of extraneous proteins such as albumin. The choice of replacement product reflects both safety issues of pathogen transmission or inhibitor development, and personal preferences of the patient and the physician. In general, currently available products are viral pathogen-free, although there is debate about the risk of transmission of parvovirus B19 and prion pathogens. Because of this very small risk, recombinant factor is the treatment of choice in previously untreated patients. In addition, a subset of concentrates contain factor that is activated during manufacture, yielding activated products that can be used in the treatment of patients with inhibitors. Such activated products, especially recombinant factor VIIa (rFVIIa), have also acquired several off-label indications in the management of bleeding in non-hemophiliac patients. The management of hemophilia patients with inhibitors is an ongoing challenge. Immune tolerance induction using a desensitization technique is successful in up to 90% of patients with alloantibodies against factor VIII, with greatest success seen in patients with low titer inhibitors who are treated soon after detection of an alloantibody and in whom treatment includes administration of immunosuppression along with repeated infusions of high titer concentrates. Such therapy is less successful in patients with factor IX alloantibodies. Non-hemophiliac patients with acquired inhibitors represent a unique patient population that requires special management. These patients have a mortality rate that approaches 25% because of the association of acquired inhibitors with severe bleeding complications, occurrence in a largely elderly population, and the frequent presence of an underlying, often serious, primary medical condition. Treatment consists of immunosuppression with steroids, chemotherapy, or intravenous immunoglobulin. Recent studies using rituximab for selective B-cell depletion in these patients have been very promising, although prospective controlled studies have not yet been performed. Finally, although hemophilia A and B appear to be ideal diseases to target with gene therapy approaches, the promise of this therapy remains to be realized.
Product Options for Replacement Therapy
The major precept of hemophilia care consists of adequate replacement of the deficient coagulation factor protein so as to prevent or reverse acute bleeding episodes. This is most effectively and efficiently accomplished by the administration of clotting factor concentrates, which contain an abundance of the specific deficient coagulation factor. Currently in the United States a multiple varieties of standard and modified factor VIII (FVIII), factor IX (FIX), and recombinant activated factor VII (rFVIIa) concentrates are available for the hemophilias and are categorized according to: (1) their source material, e.g., pooled normal plasma versus genetically engineered in “perpetual” mammalian cell lines; (2) their degree of purity, e.g., calculated on the basis of their specific activity (International Units [IU] of specific clotting factor activity/mg of total protein); (3) the viral pathogen inactivation methods employed during manufacture, e.g. heat treatment, addition of solvent detergents, chromatographic separation steps, and nanofiltration, or combinations of the above; (4) by whether they have been “activated” during manufacture, e.g., activated prothrombin complex concentrates (APCCs) and rFVIIa (used in allo- and autoantibody inhibitor patients) versus non-activated PCCs (used for hemophilia B or low titer factor IX inhibitors); and (5) finally, for rFVIIa, rFVIII, and rFIX, by the presence or absence of extraneous animal proteins or human albumin in the cell culture milieu as a nutrient source or in the final product as a stabilizer, e.g., first generation FVIII products contain human and animal albumin in both the cell culture and the final product whereas third generation products have no animal or human protein present (except for the specific purified, recombinant human clotting factor protein) at any stage of production.
The choice of which replacement product to use is determined primarily by perception of safety from pathogen transmission and from alloantibody inhibitor development; however, other variables, including patient and/or physician preference, cost and reimbursement exigencies, product availability, and, interestingly, patient “value-added” features such as convenient vial sizes, innovative syringe delivery systems, etc., all contribute to the final decision. Fortunately, it is accepted that all of the currently available replacement products are equally effective for the treatment and prophylaxis of bleeding events (exclusive of inhibitors) and it is generally recognized that all products are virtually viral-safe. What is not so clear is what the tolerance of the patient or physician is to the extremely low but theoretical potential for transmission of nonlipid-enveloped pathogens, e.g., parvovirus B19, or prions, e.g., variant Creutzfeld-Jakob disease (vCJD), in plasma-derived products or the theoretical risk of pathogen transmission for the use of hamster cell cultures for the recombinant products (Table 1 ). This also has become a major problem for individuals with von Willebrand disease, who are dependent on intermediate purity FVIII concentrates, some of which contain functional von Willebrand factor protein. No transmission of human immunodeficiency virus (HIV), hepatitis C virus (HCV), hepatitis B virus (HBV), or hepatitis A virus (HAV) has been documented for any replacement products licensed in the United States since the mid-1980s and no cases of vCJD have ever been reported anywhere with the use of any factor concentrate; however, because vCJD occurred in the UK in a recipient of packed red blood cells from a donor who was subsequently identified as being infected and because a plasma-derived FVIII concentrate was recalled recently in France because of a vCJD infected donor, recombinant products generally are perceived as potentially safer than plasma-derived products. There is general consensus that previously untreated patients with hemophilia should receive recombinant clotting factor concentrates, if at all possible. In the US, over 90% of clotting factor replacement therapy is recombinant in nature. These subtleties in the safety and purity of replacement products are particularly limited to patients in more developed countries since over 80% of the world’s hemophilia patients receive inadequate or no replacement therapy.
Special mention should be devoted to several of the newer replacement products introduced to the market. rFVIIa concentrate is licensed in the US for treatment and prevention of bleeding in hemophilia patients with alloantibody inhibitors to FVIII or FIX.1 In Europe, rFVIIa is also approved for replacement therapy for FVII deficiency. A proposed mechanism of action of rFVIIa (Figure 1 ) has provided insight into the critical role of tissue factor-FVIIa and platelet surface interactions to trigger thrombin generation through the intrinsic pathway of the coagulation cascade. The same arguments for theoretically improved pathogen safety exist for rFVIIa versus the plasma derived bypassing agents, APCC (FEIBA) and PCCs, for treatment of high titer inhibitors. In contrast to the plasma-derived bypassing agents, rFVIIa does not stimulate anamnestic responses in inhibitor patients (which may complicate the achievement of adequate coagulation) and would not be associated with the risk of severe anaphylaxis and nephrotic syndrome in those who have developed FIX inhibitors. There has been concern and controversy that rFVIIa may be more thrombogenic than plasma-derived bypassing agents, but no head-to-head comparative studies have been conducted. A recent review of data from the MedWatch pharmacovigilance program of the US Food and Drug Administration (FDA)11 suggested that thrombotic adverse events were significantly more frequent in rFVIIa than recipients of activated PCC’s (FEIBA) (incidence rate ratio, 2.98; CI, 1.71–5.52); however, this conclusion was potentially flawed by the voluntary nature of reporting these adverse events to the FDA registry with limited confirmatory information required to establish actual cause and effect. In addition, the likelihood of reporting an adverse event associated with a new medication, e.g., rFVIIa concentrate, in the marketplace is much greater than the likelihood of reporting adverse events associated with a product well established in the marketplace. Finally, the Medwatch database contained very few patients with hemophilia-related inhibitors and reflected the very widespread use of rFVIIa for “off label” high risk clinical situations with inherent thrombotic potential. The thrombosis incidence of all bypassing agents, whether plasma derived or recombinant, is extremely low in hemophilia patients so that the benefits far outweigh the risks of their administration.
A variation on the theme of genetically engineered FVIII concentrates is the second generation B-domain deleted (BDD) rFVIII (moroctocog alpha, ReFacto, Wyeth), which is formulated without human serum albumin. The unique aspect of this product is the absence of the B-domain, which is not needed for clotting activity. This results in a rFVIII molecule of 170 kDa, compared to full-length wild type rFVIII of approximately 280 kDa. After proteolytic cleavage by thrombin the activated B-domain–deleted molecule is essentially identical to the structure of activated full-length, native rFVIII. The synthesis of BDD-rFVIII is increased 15-fold by transfected cell lines compared to native wild type rFVIII; however, its efficient secretion is hindered by defective interactions with chaperone proteins in the endoplasmic reticulum necessary for transport of a properly folded molecule to the Golgi apparatus. BDD-rFVIII constructs are being exploited in the production of new FVIII products and in gene therapy. One of the problems which has emerged with BDD-rFVIII concentrates and which has been of practical concern clinically is the fact that one-stage FVIII coagulation assays employing reagents used in aPTT assays often result in about a 50% lower FVIII activity than observed with chromogenic assays. This discrepancy is also present in ex vivo plasma samples assayed after treatment with rFVIII and has been attributed to decreased phospholipid concentration. Assay discrepancies disappeared when the composition of phospholipid used in the one-stage assay was changed to contain less than 10% phosphatidylserine compared to phosphatidylcholine. For clinical monitoring, a modified BDD rFVIII standard has been developed to eliminate the assay discrepancies. Essentially, the manufacturer has reassigned the potency of its usual standard at a level 20% higher than originally and has increased the amount of protein in each vial of BDD rFVIII by 20%; however, the marketed labeled dosage will remain unchanged.2
There was initially concern that a modified molecule such as BDD-FVIII could present neoantigens and trigger an immune response in the host recipient to form a neutralizing alloantibody inhibitor. In fact, comparison of the occurrence of inhibitors for all recombinant FVIII replacement concentrates in both previously untreated (PUPs) and previously treated (PTPs) patients demonstrates similar FVIII inhibitor observed incidence across the spectrum of rFVIII products. Approximately 30% of all PUPs and up to 3% of PTPs will develop alloantibody FVIII inhibitors with the use of rFVIII products. This incidence is comparable to that observed in hemophiliacs who received multiple plasma derived factor concentrates.3,4
In an attempt to reduce the potential for recombinant replacement products to transmit blood-borne pathogens such as prions in fetal calf serum added to the media for growth of transfected cell lines or murine viruses contained in the monoclonal antibodies employed in the purification techniques, manufacturing techniques have been devised to produce concentrates free of human or serum albumin and free of human and mammalian derived nutrients or stabilizers in the synthetic process. Kogenate® Bayer/Helixate® NexGen (full length rFVIII) and ReFacto (BDD rFVIII, Wyeth) were licensed in the US in 2000 as second generation rFVIII by virtue of their human serum albumin–free final formulations. In 2003 Advate® (full length rFVIII, Baxter) received designation by the FDA as the first third generation rFVIII product since no human or mammalian derived raw materials were used in the synthetic process and the final formulation was free of human and bovine serum albumin. A third generation ReFacto has been developed and its approval is pending results of clinical trials. BeneFIX, the only available rFIX, is also synthesized in a cell system with no added mammalian proteins, and is free of extraneous albumin in the final formulation.
Future rFVIII and rFIX constructs will include novel molecules intended to: (1) increase the synthetic yield of the recombinant protein by the transfected cell culture systems; (2) prolong the circulating t½ in plasma; (3) synthesize the recombinant protein in the breast milk of a transgenic animal; and (4) modify the epitope regions on the A1, A2, and C2 domains of rFVIII to render the molecule less antigenic.5
Immune Tolerance Induction
Immune tolerance induction (ITI) is an immune system “desensitization” technique intended to eradicate an alloantibody inhibitor, which neutralizes a specific coagulation factor function.6 These inhibitors develop in up to 30% of individuals with severe hemophilia A and 3% with hemophilia B. Typically, daily or bid FVIII or FIX concentrates are administered until the inhibitor titer is no longer detectable in the Bethesda Unit laboratory assay and the clotting factor recovery and circulating t½ in plasma are normalized. Bypassing agents (Table 1 ) should be used to treat or prevent acute bleeding complications, which may become more frequent as anamnestic responses to ITI occur. Some ITI regimens are complemented by administration of immunosuppressive medications and/or extracorporeal plasmapheresis and/or immunoadsorption to reduce the inhibitor titers and thus improve the chances of ITI success. The success of ITI approaches 90% usually over approximately 6–12 months for alloFVIII antibody inhibitors and the following variables appear to be good prognostic indicators7:
Initiation of ITI soon after detection of the alloantibody
Initiation of ITI after reduction of antibody titer to a low level employing immunosuppression or mechanical means or waiting for the titer to decay from high to low level
Low titer, low responder (non-anamnestic) inhibitor (< 5 Bethesda Units)
Use of plasma derived FVIII concentrates rich in von Willebrand factor
Use of FVIII concentrates in amounts large enough (200 IU/kg/d) to achieve detectable FVIII activity in laboratory assays (for high titer inhibitors); low titer inhibitors may be suppressed with lower amounts of FVIII (25 IU/kg/d)
A large prospective randomized clinical trial is in progress to establish the validity of these variables. Once ITI is successfully achieved, a prolonged prophylaxis regimen with FVIII administered three times weekly should be instituted to consolidate inhibitor eradication.
ITI is less successful for FIX alloantibody inhibitors and may be associated with the development of severe anaphylaxis and nephrotic syndrome. Patients should be informed about the possibility of anaphylaxis if ITI is attempted in this setting. These individuals often have large FIX gene deletions and require rFVIIa to treat their acute bleeding episodes so as to avoid further exposure to any additional FIX antigen.
Acquired Autoantibody Inhibitors (Acquired Hemophilia)
Autoantibody inhibitors, predominantly targeting FVIII in individuals with previously normal coagulation, occur with an estimated incidence of 1–3 per million population per year. The mortality rate associated with acquired autoantibody inhibitors approaches 25% versus the substantially lower risk of death in those with alloantibodies. Compared to alloantibody inhibitor patients, acquired hemophilia is characterized by: (1) a more severe bleeding pattern; (2) higher incidence in older population; (3) occurrence in conjunction with identifiable underlying autoimmune diseases, lymphoproliferative or solid tumor malignancies, pregnancy, and use of certain antibiotics such as penicillin and sulfonamides in approximately 50% of cases; and (4) in vitro inhibitor activity that follow a type II pharmacokinetic pattern with incomplete neutralization of the targeted clotting factor activity by the autoantibody, typically resulting in residual factor VIII levels ranging between 2%–18% in patient plasma. In some, the aPTT is only mildly prolonged or in the upper normal range.
Allo- and autoantibodies bind to the FVIII molecule are polyclonal with remarkable fidelity to immunodominant epitopes on the A2 and C2 domains and a minor epitope on A3. The epitope targets for the allo and auto-FVIII antibody inhibitors overlap with FVIII binding sites for phospholipids, von Willebrand factor protein, factor X, and FIX. The majority of auto-FVIII:C inhibitory antibodies (62%) are predominantly directed against epitopes on either the C2 or A2 domains; whereas interactions by allo-FVIII:C inhibitors much more commonly involved species that were strongly reactive to epitopes in both domains (85%) in hemophilic plasmas. In addition, anti-A2 domain targeted inhibitors predominate in allo-FVIII:C antibody inhibitor plasmas (71%) but are detected in only a third of autoantibody plasmas.8,12
The therapy of acquired autoantibody inhibitors is based primarily on the need to control or prevent acute hemorrhagic complications, which frequently are life and limb threatening and secondarily to eradicate the autoantibody to restore normal coagulation. While most bleeds associated with low titer autoantibody inhibitors (< 5 Bethesda Units) may be treated effectively with FVIII concentrates administered at high doses, the correlation between Bethesda Unit titer and response to therapy is not as predictable as it is with alloantibody inhibitors. Figure 2 provides a treatment algorithm for autoantibody inhibitor related bleeding.
Porcine FVIII concentrate was formerly considered a critical first-line therapy for acquired hemophilia-related bleeding since it was the only replacement therapy that provided an opportunity to actually measure post-infusion FVIII coagulation activity levels in the laboratory. The product was removed from the marketplace in 2004 because of contamination of the porcine plasma pools by porcine parvovirus. There is a recombinant form of porcine FVIII concentrate currently in clinical trials, but clinical efficacy has not yet been established. Thus, “bypassing” agents (Table 1 ) are most commonly used, but potential risks of thrombogenicity exist and there is only about 80% efficacy for each product. Plasma exchange via plasmapheresis and extracorporeal immunoadsorption may be necessary to temporarily reduce the inhibitor titer enough for bypassing agents or FVIII replacement to provide adequate hemostasis.
Eradication of autoantibody inhibitors depends on immunosuppressive measures, such as: (1) administration of corticosteroids with 30%–50% efficacy in 3–6 weeks; (2) use of cytotoxic and myelosuppressive chemotherapeutic agents, e.g., cyclophosphamide, cyclosporine, 2-chlorodeoxyadenosine; (3) immunomodulation with intravenous immunoglobulin; and (4) selective B-lymphocyte depletion with rituximab. This latter approach is very promising, but prospective and controlled studies remain to be conducted. The exact efficacy of rituximab has not been established; despite early reports of dramatic responses, subsequent failures have also been reported. Rituximab responders may require concurrent use of steroids and relapses may respond to retreatment.9 Recent immune tolerance induction regimens have successfully and rapidly eradicated the autoantibody inhibitor and this approach may emerge as preferable first-line therapy.10
GeneTherapies
Hemophilia A and B are ideal disease states to target for gene therapy since they are caused by mutations in single identified genes, a slight increase in clotting factor levels in vivo can convert severe hemophilia into milder disease, and current replacement therapies will always be considered suboptimal. Also, there is a wide range of safety if there is an “overshoot” of desired level of coagulation activity. Unfortunately, to date the promise of gene therapy and a cure for the hemophilia patient have not been realized, primarily because a gene delivery system which is non-immunogenic enough to allow for long term expression of the clotting factor activity has not been achieved completely. Furthermore, strategies for gene therapy in general have been modified following a death experienced in a gene therapy trial for ornithine transcarbamylase deficiency, and due to cases of acute T-cell leukemia reported in X-linked recessive SCID treated with retroviral vectors, attributable to insertional mutagenesis. Despite the relative safety and some indications of prolonged expression gained from the 6 trials conducted in the hemophilias (Table 2 ), there are currently no gene therapy trials open for hemophilia, although additional trials are under review. If specific FVIII or FIX gene therapy cannot be immediately achieved, then other novel therapeutic approaches may offer alternative benefits, including: (1) the use of gene delivery of engineered secreted, activated FVII; (2) applications of new viral vector technology; (3) use of nanoparticle technology for the delivery of genes to hepatocytes; or (4) gene “pharming.” Any of these, if successful, could enhance the quality of life for the individual with hemophilia and potentially offer an approach to treatment of patients in developing countries where concentrate therapy is unavailable.
Clotting factor concentrates available in the United States in 2005.
| Product Name (Manufacturer) | Viral Inactivation Procedure(s) | Purity/Specific Activity (IU Factor VIII activity/mg total protein) Before Addition of Stabilizer |
|---|---|---|
| Human Plasma-derived Factor VIII Concentrates | ||
| Humate-P (ZLB Behring, Inc. ) (Contains functional von Willebrand factor protein) | Pasteurization (heating in solution, 60°C, 10 hr) | Intermediate (1–10 IU/mg) |
| Alphanate SD (Grifols, Inc.) (Contains some functional von Willebrand factor protein) | Solvent detergent (TNBP/Polysorbate 80) Affinity chromatography Dry heat (80°C, 72 hr) | High (50–100 IU/mg) (> 400 IU/mg after correcting for VWF content) |
| Koate-DVI (Bayer, Inc.) (Contains von Willebrand factor protein) | Solvent detergent (TNBP/Polysorbate 80) Dry heat (80°C, 72 hr) | High (50–100 IU/mg) |
| Monoclonal Antibody Purified FVIII Concentrates (Immunoaffinity purified from human plasma, no intact von Willebrand factor protein) | ||
| Monarc-M (Baxter/Immuno, Inc., using recovered plasma from the American Red Cross) | Solvent detergent (TNBP/Octoxynol 9) Immunoaffinity chromatography | Ultra high (> 3000 IU/mg) |
| Hemofil-M (Baxter/Immuno, Inc.) | Solvent detergent (TNBP/Octoxynol 9) Immunoaffinity chromatography | Ultra high (> 3000 IU/mg) |
| Monoclate-P (ZLB Behring, Inc.) | Pasteurization (heated in solution, 60°C, 10 hr) Immunoaffinity chromatography | Ultra high (> 3000 IU/mg) |
| Recombinant (Genetic Engineered)/First Generation FVIII Concentrates | ||
| Recombinate (Baxter/Immuno, Inc.) (human albumin as a stabilizer) | Immunoaffinity, Ion exchange chromatography Bovine serum albumin used in culture medium for Chinese hamster ovary cells | Ultra high (> 4000 IU/mg) |
| Recombinant/Second Generation FVIII Concentrates (human albumin-free final formulations) | ||
| Kogenate FS (Bayer, Inc.) Helixate FS (Bayer for ZLB Behring, Inc.) (sucrose as a stabilizer) | Immunoaffinity chromatography Ion exchange Solvent detergent (TNBP/polysorbate 80) Ultrafiltration | Ultra high (> 4000 IU/mg) |
| Refacto (Wyeth, Inc.) B-domain deleted (sucrose as a stabilizer) | Ion exchange Solvent detergent (TNBP/Triton X-100) Nanofiltration | Ultra high (> 11,200–15,500 IU/mg), measured via chromogenic assay technique |
| Recombinant/Third Generation FVIII Concentrates (No human or animal protein used in the culture medium or manufacturing process; does contain trace amounts of murine monoclonal antibody) | ||
| Advate (Baxter/Immuno, Inc.) (trehalose as a stabilizer) | Immunoaffinity chromatography Ion exchange Solvent detergent (TNBP/polysorbate 80) | Ultra high (> 4000–10,000 IU/mg) |
| Plasma-derived Prothrombin Complex Concentrates/Factor IX complex concentrates (nonactivated; also contain FX & prothrombin but only traces of FVII ) | ||
| Bebulin VH (Baxter/Immuno) | Vapor heat (60°C, 10 hr at 190 mbar pressure plus 1 hr at 80°C, 375 mbar) | Intermediate (< 50 IU/mg) |
| Profilnine SD (Grifols) | Solvent detergent (TNBP/polysorbate 80) | Intermediate (< 50 IU/mg) |
| Proplex-T (Baxter/Immuno) | Dry heat (60°C, 144 hr) | Intermediate (< 50 IU/mg) |
| Plasma-derived Prothrombin Complex Concentrates/Factor IX complex concentrates (activated) | ||
| FEIBA (Baxter/Immuno) | Vapor heating (60°C, 10 hr, 1160 mbar) | Intermediate (< 50 U/mg) |
| Plasma-derived Coagulation Factor IX (human) concentrates | ||
| AlphaNine SD (Grifols) | Dual affinity chromatography Solvent detergent (TNBP/polysorbate 80) Nanofiltration (viral filter) | High (> 200 IU/mg) |
| Mononine (ZLB Behring) | Monoclonal antibody immunoaffinity chromatography Sodium thiocyanate Ultrafiltration | High (> 160 IU/mg) |
| Recombinant F.IX Concentrates | ||
| BeneFIX (Wyeth) (no animal or human-derived protein in cell line; no albumin added to final product) | Affinity chromatography Ultrafiltration | Ultra-high (> 200 IU/mg) |
| Product Name (Manufacturer) | Viral Inactivation Procedure(s) | Purity/Specific Activity (IU Factor VIII activity/mg total protein) Before Addition of Stabilizer |
|---|---|---|
| Human Plasma-derived Factor VIII Concentrates | ||
| Humate-P (ZLB Behring, Inc. ) (Contains functional von Willebrand factor protein) | Pasteurization (heating in solution, 60°C, 10 hr) | Intermediate (1–10 IU/mg) |
| Alphanate SD (Grifols, Inc.) (Contains some functional von Willebrand factor protein) | Solvent detergent (TNBP/Polysorbate 80) Affinity chromatography Dry heat (80°C, 72 hr) | High (50–100 IU/mg) (> 400 IU/mg after correcting for VWF content) |
| Koate-DVI (Bayer, Inc.) (Contains von Willebrand factor protein) | Solvent detergent (TNBP/Polysorbate 80) Dry heat (80°C, 72 hr) | High (50–100 IU/mg) |
| Monoclonal Antibody Purified FVIII Concentrates (Immunoaffinity purified from human plasma, no intact von Willebrand factor protein) | ||
| Monarc-M (Baxter/Immuno, Inc., using recovered plasma from the American Red Cross) | Solvent detergent (TNBP/Octoxynol 9) Immunoaffinity chromatography | Ultra high (> 3000 IU/mg) |
| Hemofil-M (Baxter/Immuno, Inc.) | Solvent detergent (TNBP/Octoxynol 9) Immunoaffinity chromatography | Ultra high (> 3000 IU/mg) |
| Monoclate-P (ZLB Behring, Inc.) | Pasteurization (heated in solution, 60°C, 10 hr) Immunoaffinity chromatography | Ultra high (> 3000 IU/mg) |
| Recombinant (Genetic Engineered)/First Generation FVIII Concentrates | ||
| Recombinate (Baxter/Immuno, Inc.) (human albumin as a stabilizer) | Immunoaffinity, Ion exchange chromatography Bovine serum albumin used in culture medium for Chinese hamster ovary cells | Ultra high (> 4000 IU/mg) |
| Recombinant/Second Generation FVIII Concentrates (human albumin-free final formulations) | ||
| Kogenate FS (Bayer, Inc.) Helixate FS (Bayer for ZLB Behring, Inc.) (sucrose as a stabilizer) | Immunoaffinity chromatography Ion exchange Solvent detergent (TNBP/polysorbate 80) Ultrafiltration | Ultra high (> 4000 IU/mg) |
| Refacto (Wyeth, Inc.) B-domain deleted (sucrose as a stabilizer) | Ion exchange Solvent detergent (TNBP/Triton X-100) Nanofiltration | Ultra high (> 11,200–15,500 IU/mg), measured via chromogenic assay technique |
| Recombinant/Third Generation FVIII Concentrates (No human or animal protein used in the culture medium or manufacturing process; does contain trace amounts of murine monoclonal antibody) | ||
| Advate (Baxter/Immuno, Inc.) (trehalose as a stabilizer) | Immunoaffinity chromatography Ion exchange Solvent detergent (TNBP/polysorbate 80) | Ultra high (> 4000–10,000 IU/mg) |
| Plasma-derived Prothrombin Complex Concentrates/Factor IX complex concentrates (nonactivated; also contain FX & prothrombin but only traces of FVII ) | ||
| Bebulin VH (Baxter/Immuno) | Vapor heat (60°C, 10 hr at 190 mbar pressure plus 1 hr at 80°C, 375 mbar) | Intermediate (< 50 IU/mg) |
| Profilnine SD (Grifols) | Solvent detergent (TNBP/polysorbate 80) | Intermediate (< 50 IU/mg) |
| Proplex-T (Baxter/Immuno) | Dry heat (60°C, 144 hr) | Intermediate (< 50 IU/mg) |
| Plasma-derived Prothrombin Complex Concentrates/Factor IX complex concentrates (activated) | ||
| FEIBA (Baxter/Immuno) | Vapor heating (60°C, 10 hr, 1160 mbar) | Intermediate (< 50 U/mg) |
| Plasma-derived Coagulation Factor IX (human) concentrates | ||
| AlphaNine SD (Grifols) | Dual affinity chromatography Solvent detergent (TNBP/polysorbate 80) Nanofiltration (viral filter) | High (> 200 IU/mg) |
| Mononine (ZLB Behring) | Monoclonal antibody immunoaffinity chromatography Sodium thiocyanate Ultrafiltration | High (> 160 IU/mg) |
| Recombinant F.IX Concentrates | ||
| BeneFIX (Wyeth) (no animal or human-derived protein in cell line; no albumin added to final product) | Affinity chromatography Ultrafiltration | Ultra-high (> 200 IU/mg) |
| Factor VIII (and/or Factor IX) Concentrates Useful in Treatment of Alloantibody and Autoantibody Inhibitor Related Bleeding | ||
|---|---|---|
| Product Name (Manufacturer) | Viral Inactivation Method | Dosage |
| Recombinant factor VIIa (Genetically engineered) | ||
| NovoSeven (Novo Nordisk, Inc.) (Stabilized in mannitol; bovine calf serum used in culture medium) | Affinity chromatography Solvent/detergent (TNPB/polysorbate 80) | 90 μg/Kg intravenous bolus every 2–3 hr until bleeding ceases (larger dosing regimens are experimental but may be useful in refractory bleeding). This product is the treatment of choice for individuals with allo-factor IX antibody inhibitors and anaphylaxis and/or renal disease associated with the use of factor IX containing concentrates |
| FEIBA-VH (Baxter Immuno, Inc.) (human plasma-derived) | Vapor heated (10 hr, 60°C, 190 mbar plus 1 hr, 80° C, 375 mbar) | 50–100 IU/kg not to exceed 200 IU/kg/24 hr (for factor VIII and IX inhibitors) |
| Porcine Plasma-derived Factor VIII Concentrate | ||
| Hyate C (Ibsen Biomeasure, Inc.) | No longer available | > 50 IU/mg (for factor VIII inhibitors only) |
| Factor VIII (and/or Factor IX) Concentrates Useful in Treatment of Alloantibody and Autoantibody Inhibitor Related Bleeding | ||
|---|---|---|
| Product Name (Manufacturer) | Viral Inactivation Method | Dosage |
| Recombinant factor VIIa (Genetically engineered) | ||
| NovoSeven (Novo Nordisk, Inc.) (Stabilized in mannitol; bovine calf serum used in culture medium) | Affinity chromatography Solvent/detergent (TNPB/polysorbate 80) | 90 μg/Kg intravenous bolus every 2–3 hr until bleeding ceases (larger dosing regimens are experimental but may be useful in refractory bleeding). This product is the treatment of choice for individuals with allo-factor IX antibody inhibitors and anaphylaxis and/or renal disease associated with the use of factor IX containing concentrates |
| FEIBA-VH (Baxter Immuno, Inc.) (human plasma-derived) | Vapor heated (10 hr, 60°C, 190 mbar plus 1 hr, 80° C, 375 mbar) | 50–100 IU/kg not to exceed 200 IU/kg/24 hr (for factor VIII and IX inhibitors) |
| Porcine Plasma-derived Factor VIII Concentrate | ||
| Hyate C (Ibsen Biomeasure, Inc.) | No longer available | > 50 IU/mg (for factor VIII inhibitors only) |
Summary of clinical trials of gene transfer for hemophilia.
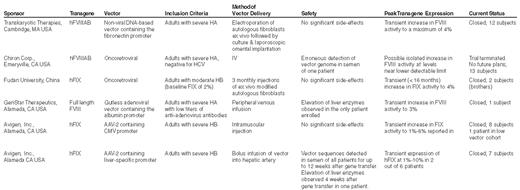
Abbreviations: hFVIIIAB, B-domain-deleted factor FVIII; HA, hemophilia A; hFIX, human FIX; HB, hemophilia B; CMV, cytomegalovirus; HCV, hepatitis C virus. Updated and modified from Table IV in: Nathwani,

A cell-based model of coagulation (see text for further explanation). Adapted from Jurlander B. Seminars in Thrombosis and Haemostasis. 27:373,2001. This model also is amenable to a tissue factor independent mechanism of thrombin generation in the absence of tissue factor and FVIII or FIX, such as would be the condition in severe congenital hemophilia A or B with alloantibody inhibitors or in acquired hemophilia with autoantibody inhibitors. In these situations According to the findings of Monroe et al.13 suprapharmacologic doses of rFVIIa can bind non-specifically to the activated platelet without the requirement of TF and subsequently can activate factor X to Xa in the absence of factor VIII or IX. In contrast, the Mann model14 suggests that suprapharmacologic doses of rFVIIa activates coagulation by overcoming an intrinsic inhibition of zymogen FVII in the absence of TF.
A cell-based model of coagulation (see text for further explanation). Adapted from Jurlander B. Seminars in Thrombosis and Haemostasis. 27:373,2001. This model also is amenable to a tissue factor independent mechanism of thrombin generation in the absence of tissue factor and FVIII or FIX, such as would be the condition in severe congenital hemophilia A or B with alloantibody inhibitors or in acquired hemophilia with autoantibody inhibitors. In these situations According to the findings of Monroe et al.13 suprapharmacologic doses of rFVIIa can bind non-specifically to the activated platelet without the requirement of TF and subsequently can activate factor X to Xa in the absence of factor VIII or IX. In contrast, the Mann model14 suggests that suprapharmacologic doses of rFVIIa activates coagulation by overcoming an intrinsic inhibition of zymogen FVII in the absence of TF.

