
Abstract
The pathogenesis of acute myelogenous leukemia (AML) involves an array of molecular alterations that disrupt almost every facet of cell transformation. These processes include the regulation of cell proliferation, differentiation, self-renewal, survival, cell cycle checkpoint control, DNA repair and chromatin stability, and cell dissemination. Normal regulatory networks are disrupted or usurped by these leukemogenic insults, and the understanding of these alterations is guiding the design of new therapeutic strategies. This overview describes some of the critical molecular alterations and implicates the rogue leukemogenic proteins in the onset and progression of AML.
Insight into the molecular basis of acute myelogenous leukemia (AML) may come from a reductionist approach, in which the characteristics of the leukemic cell are correlated with the genetic lesions that cause them. Some of these features of AML and their potential causes are listed in Table 1 . These include inappropriate proliferation in the absence of normal growth signals, indefinite self-renewal in a manner analogous to a stem cell, escape from programmed cell death, inhibition of differentiation, aberrant cell cycle checkpoint control, genomic instability and multi-organ dissemination of leukemic cells. These properties may be directly linked to focal genetic lesions in AML and represent discrete processes amenable to therapy. However, from the perspective of systems biology it is clear that many of these properties are intertwined and result from integration of multiple anomalies in the genetic program of the leukemic cell.
Inappropriate Proliferation: The Role of Signaling Molecules
Activation of receptor and intracellular protein tyrosine kinases stimulates a cascade of phosphorylation-driven protein docking and recruitment events that leads to the alteration of transcription in the cell nucleus and the stimulation of cell cycle progression. Although cell proliferation is regulated by the presence of growth factors and adhesion signals in normal cells, it can be triggered in leukemic cells in a cell autonomous manner. This abnormal proliferation is often the result of mutations affecting proliferative signaling pathways. Following the discovery of the BCR-ABL tyrosine kinase in chronic myelogenous leukemia, other activated kinases have become causally implicated in the pathogenesis of AML. The FLT3 tyrosine kinase is expressed in almost all patients with AML. It is constitutively activated by internal tandem duplication within the juxtamembrane domain or by mutation within the activation loop of the kinase in approximately 30% of AML. This finding has stimulated clinical trials of FLT3 inhibitors, yielding some clinical responses (reviewed in 1, and see also section below on “New treatment strategies…”).
The c-KIT tyrosine kinase is expressed in 60%–80% of AML patients, and this kinase is activated by mutation in mast cell leukemia and some cases of AML. The JAK2 kinase is activated by the V617F point mutation in the overwhelming majority of patients with polycythemia vera and in a significant proportion of patients with essential thrombocythemia or idiopathic myelofibrosis (reviewed in 2), all disorders that can evolve into AML. Activated JAK2 induces proliferation in part through engagement of the STAT family of transcriptional activators. The V617F JAK2 mutation is found in 5% of patients with myelodysplastic syndrome,3 suggesting that the mutation will also be found in patients who transform to frank AML.
Activated tyrosine kinases can transmit proliferative signals by engagement of the RAS family of small G-proteins, and mutation of genes encoding these proteins can mimic the effects of receptor tyrosine kinase (RTK) mutations. NRAS is mutated and constitutively activated in 10%–20% of AML, KRAS in 5%–15% of patients, while HRAS is rarely mutated.4 AML samples that contain RAS mutations do not have kinase fusions or activating mutations, suggesting that Ras and tyrosine kinase molecules fall within a single complementation group in AML. In general the RTK pathways in AML are activated by gain of function mutations. The exception is the loss of expression of the tumor suppressor protein neurofibromin (NF1), which inactivates RAS by enhancing its intrinsic GTPase activity. Children with neurofibromatosis have a germline loss of one NF1 allele and can develop juvenile myelomonocytic leukemia (JMML) or AML with loss of the remaining normal NF1 allele. However, NF1 loss is rare in de novo childhood and adult AML. Another way to activate the RTK pathway is by activation of the phosphatase activity of SHP-2/PTPN11 through point mutation. Although seemingly paradoxical, the removal of phosphate groups from certain key substrates stimulates signaling through RTK pathways. Initially described in JMML, a small proportion of patients with de novo AML have SHP-2 activating mutations.5 RTK pathway mutations have been documented in nearly 50% of cases of AML. It can be postulated that all cases of AML are associated with a genetic abnormality leading to deregulation of proliferative signaling. Genome sequencing efforts are underway at multiple centers to identify these mutations.
The characterization of these signaling pathway abnormalities has focused attention on cell proliferation as a potential therapeutic target in AML. The obstacle to this strategy is the multiple ways by which kinase signaling may be activated in AML. It remains to be determined whether kinase-directed therapy needs to be personalized to each patient based upon the unique profile of mutations or will be robust enough to interfere with the signaling pathway irrespective of the specific lesion.
Differentiation Blockade: The Role of Transcription Factors in AML
Transcription factors are commonly disrupted in AML either by their fusion as a result of chromosomal translocation or by point mutation. Factors affected by chromosomal rearrangement include the core binding factor complex, the retinoic acid receptor (RAR), the MLL protein, and Hox proteins. Point mutations in myeloid transcription factors, including C/EBPα and PU.1, may also lead to loss of normal myeloid differentiation in AML. Chimeric transcription factors often work as dominant negative forms of the original factor. CBF and RAR fusions are prime examples of this.
The core binding factor complex consists of a heterodimer of the Runx1 (formerly AML1) and CFBβ protein and normally activates a number of genes critical for normal myeloid development. In AML this complex factor is disrupted by at least three different translocations: t(8;21), which generates the Runx1-MTG8 fusion; inversion of chromosome 16, yielding the CBFβ-MYH11 fusion; and t(3,21), which generates the RUNX1-EVI1 fusion protein associated with MDS and therapy-related AML. All of these fusions act at least in part as dominant negative forms of the core binding factor complex. In addition, point mutations in the DNA binding domain of Runx1 are associated with a familial platelet disorder with predisposition to leukemia, as well as with rare cases of sporadic AML. These findings suggest that loss of RUNX1 function can lead to deregulated differentiation and leukemia. RUNX1-MTG8 represses genes usually activated by RUNX1, including c-FMS,6 p14 ARF,7 and C/EBPα 8 through recruitment of co-repressor complexes. This is due to the fact that MTG8 is a transcriptional co-repressor that binds histone deacetylases, other co-repressors and transcription factors. DNA methyl transferases may also be recruited to RUNX1 target genes, potentially leading to longer lasting epigenetic silencing of target genes.9 Repression of Runx1 target genes acts to inhibit the normal differentiation program. Repression of p14 ARF may functionally destabilize p53 and allow other mutations to accumulate in the leukemic cell. Treatment with inhibitors of histone deacetylase can induce differentiation of cell lines harboring RUNX1/MTG8.10 Alternatively, expression of a decoy peptide that blocks the interaction of co-repressors with the Runx1/MTG8 oncoprotein can also reverse transcriptional repression and allow cell differentiation to proceed.10
RUNX1-MTG8 and other fusion oncoproteins also activate genes critical for leukemia pathogenesis. When expressed in human CD34+ cells, RUNX1-MTG8 allows for the continuous self-renewal of immature cells.11 Similarly, when expressed in murine marrow RUNX1-MTG8 allows for the serial replating of marrow progenitor cells ex vivo.12 This observation can be correlated with the finding that RUNX1-MTG8 as well as the PML-RARα fusion protein described below can activate the expression of components of the Wnt signaling pathway13,14 that promote stem cell renewal.15 Runx1-MTG8 on its own is insufficient to generate AML; however, co-expression of Runx1-MTG8 along with an activated tyrosine kinase in murine marrow (TEL-PDGFRβ ) efficiently induces myeloid leukemia.16 However a recent paper showed the surprising result that a spontaneous mutation in RUNX1-MTG8 expressing mice can lead to the formation of a truncated version of the fusion protein that efficiently induces leukemia.17 This suggests that a targeted mutation of the RTK pathway might not always be necessary for AML pathogenesis. For example, RUNX1-MTG8 upregulates Trk-A, a nerve growth factor receptor, and sensitizes cells to NGF and related growth factors, which may induce proliferation independent of RTK pathway mutation.18 Hence, although the paradigm of a transcription factor and signaling anomaly being required for AML is likely true, there may be more than one way to achieve this pairing.
Acute promyelocytic leukemia, a clear-cut example of differentiation blockade in AML, is always associated with rearrangement of the retinoic acid receptor alpha (RARα ). In over 98% of cases APL is associated with t(15;17), which generates the PML-RARα fusion protein. The PLZF-RARα fusion associated with t(11;17) represents less than 1% of cases and yields a retinoic acid–resistant form of disease. Rare fusions of RARα to STAT5b, nucleophosmin (a gene mutated in a substantial number of AML cases), as well as the NuMA nuclear matrix gene have been reported as well. Although RARα is not required for myeloid development, it acts as a molecular rheostat to regulate myeloid differentiation. Whereas normal differentiation occurs at a retinoic acid concentration of 10−8 the level present in human serum, in APL, cells are blocked at the promyelocytic stage of differentiation and self-renew. Differentiation blockade may occur as in the case of the RUNX1-MTG8 fusion by the aberrant recruitment of repression complexes to key target genes. PML and all of the fusion partner proteins in APL allow the chimeric RARα to form multimeric complexes along with RXR. One group showed that while RARα /RXR heterodimers bind to direct repeat sequences separated by 5 base pairs, PML-RARα /RXR multimers bind to direct repeat sequences separated by as many as 20 base pairs.19 Hence PML-RARα represents not just a dominant negative form of RARα but a gain of function mutation with the potential to repress an expanded repertoire of genes. In addition, differing from wild-type RARα, PML-RARα can recruit DNA methyl transferases to promoters contributing to their efficient and long-lasting suppression.20 Microarray studies comparing genes affected in cell lines engineered to express the RAR fusion proteins as well as patient specimens are beginning to define the genes repressed by the fusion proteins.21 In addition these studies have indicated that the fusion proteins also activate gene expression, most likely indirectly through competition for repressor proteins, and many of these targets are genes that promote self-renewal. As in the case of RUNX1-MTG8, expression of PML-RARα alone is able to block differentiation in cell lines but cannot do so on its own in vivo. For months before developing leukemia, transgenic mice harboring the fusion protein produce normal neutrophil that can be demonstrated to express the fusion gene. After a delay the animals develop a myeloproliferative disease and later develop frank APL. The nature of the secondary changes required for disease development and differentiation block remain uncertain. However, a recent report indicates that a specific cleavage of the PML-RARα fusion by neutrophil elastase was required for disease progression.22 In accordance with the developing model of cooperation of signaling and transcriptional pathways, FLT3 mutations are common in APL, and co-expression of mutant FLT3 and PML-RAR in mouse marrow accelerates disease development.23 Whether FLT3-mediated signaling contributes to the block to differentiation as well as to the proliferative capacity of the tumor remains to be determined. ATRA and arsenic are mainstays of treatment of APL and are thought to act by reactivation of RAR target genes and degradation of the PML-RAR fusion, leading to terminal differentiation of promyelocytes. Attempts at differentiation therapy in other forms of AML have met with limited success. The histone deacetylase inhibitor valproic acid, either alone or in combination with ATRA, has provided some preliminary evidence of differentiation and decreased blast counts in patients.24 Whether more potent histone deacetylase (HDAC) inhibitors will allow more effective release of the differentiation block in AML remains to be determined (see below).
MLL Rearrangements
MLL rearrangements offer another paradigm for deregulation of gene expression in leukemia. RUNX1-MTG8 and PML-RARα and other RARα fusion proteins are best understood as repressor proteins. MLL itself is an activator protein that through AT hook motifs can associate with specific DNA sequences, most notably in Hox gene promoters. MLL is required for Hoxc8 expression and binds and methylates histones at the Hox loci (at histone H3 lysine 4), methylation of which is associated with the activation of target genes.25 MLL-fusion proteins link the N-terminal region of the protein, which contains the DNA recognition region to one of dozens of partner proteins of diverse functions. A universal feature of the MLL-fusion proteins is their ability to dimerize with themselves and wild-type MLL. The MLL fusions appear to encourage the recruitment of MLL complexes to Hox genes to activate their expression. Hox gene expression in turn is associated with increased self-renewal by hematopoietic cells. In addition a recent report indicates that MLL-fusions prevent the acetylation and activation of p53,26 a feature that can affect cycle cycle, apoptosis and genomic stability. Some MLL fusion proteins may directly target Hox genes for activation by aberrant recruitment of activators. For example, the MLL-CBP fusion can directly target histone acetyl transferases to target genes,27 and the MLL-AF10 fusion recruits the Dotl1 histone methyl transferase to targets.28
Escape from Programmed Cell Death
The ability to evade apoptosis is critical to the development of a malignancy. Protein tyrosine kinase activation can have the dual effect of promoting cell proliferation and in addition enhancing cell survival by activating phosphatidylinositol 3-kinase (PI 3-kinase) signaling. The phospholipid products of PI 3-kinase activate the AKT serine/threonine kinase, and this kinase in turn phosphorylates BAD and releases the BCL-2 pro-survival molecule. In many instances the clinical outcome of patients with AML is correlated with altered levels of pro-apoptotic and pro-survival molecules in leukemic cells. Expression of the Bcl-2 survival molecule, the IAP family member survivin and the extrinsic death pathway protein FADD are predictive of clinical response rates and survival in AML, although why these genes have variable expression remains in large part unexplained.
The p53 protein is a focal point in the regulation of apoptotic signaling and cell cycle regulation. Mutations within p53 are associated with adverse response to chemotherapy in patients with AML. Moreover, the function of this protein is often compromised in AML as p53 regulatory molecules are usurped by leukemogenic signaling. The RUNX1-MTG8 fusion protein of AML represses the expression of p14ARF and promotes destabilization of p53. Through an alternative mechanism, the MOZ-TIF2 fusion of AML binds to CBP and indirectly attenuates the transcriptional activity of p53.29 The nucleophosmin protein (NPM), which is fused to the ALK protein tyrosine kinase in more than half of anaplastic large cell lymphoma cases, is known to regulate the stabililty of p53. Interestingly, mutations within the NPM coding region are associated with cytoplasmic localization of this protein in 35% of newly diagnosed AML.30
Self-Renewal
Unlike normal progenitor cells that are committed to a particular hematopoietic lineage, leukemic cells from patients with AML can undergo self-renewal rather than lineage-specific commitment. Moreover, the leukemic stem cell population in AML is functionally heterogeneous with differing capacities for self-renewal.31 Possible explanations for this self-renewal are beginning to emerge. As noted above, NPM is mutated in approximately one-third of newly diagnosed AML, and the expression of this cytoplasmic NPM variant is associated with expression of genes thought to support maintenance of the stem cell phenotype.32 The FLT3-ITD mutant of AML, which activates proliferative and survival pathways, also confers the property of self-renewal in human CD34+ cells.33 The Wnt/β-catenin signaling relay is a critical element in the control of self-renewal of normal and cancer stem cells. As noted above, the RUNX1-MTG8, PML-RARα , and PLZF-RARα fusions can all induce the expression of β-catenin and γ-catenin (plako-globin) proteins.13 Expression of these fusion proteins also results in expression of components of the Jagged/Notch signaling relay.21 Thus, the expression of mutated and fusion genes in AML seems to underlie some aspects of enhanced self-renewal, although such findings do not exclude the possibility that the progenitor cell in AML might itself have intrinsic self-renewal properties independent of a leukemogenic insult.
Loss of Cell Cycle Control
Deregulation of cell cycle control in AML may occur through multiple mechanisms. First, constitutive Ras/MAP kinase signaling leads to the activation of nuclear transcription factors that induce expression of cyclins. Activation of the AKT pathways can lead to degradation of the p27 CDK inhibitor. Mutation of p53, disruption of p53 function through suppression of ARF, disruption of the PML nuclear body function, or disruption of NPM ability to sequester MDM234 can all lead to decreased p53 levels and activity and the loss of G1 checkpoint control. Rb mutations and deletions occur in AML as in other forms of malignancy. Hypermethylation of growth suppressor genes is a recurrent theme in many malignancies and is well described in AML. In particular, the p15 Ink4a/ARF locus as well as the p16 Ink4b locus encoding cyclin-dependent kinase inhibitors can be methylated and silenced in AML.35 The demethylating agent azacytidine reactivates these genes in cell culture, and one study found increased p15 expression in bone marrow specimens of patients with MDS treated with demethylating agent therapy.36
Genomic Instability
The genomic instability of leukemic cells can be explained in part through the subversion of p53 by the mechanisms noted above. Defective mismatch repair has been noted in AML after solid organ and hematopoietic stem cell transplant,37 and a polymorphic splice site in MSH2 is associated with an elevated risk of AML. In addition, AML fusion transcription factors repress genes associated with DNA repair.21 The PML protein binds and stabilizes the DNA damage repair protein TopBP1. In APL, where the PML nuclear body is disrupted, TopBP1 was unable to be recruited to irradiation induced foci, presumed sites of DNA repair.38 Genomic instability in AML may also result from poor fidelity of homologous end joining activity after DNA double strand breaks.39 Collectively these defects may allow for the continued evolution of the leukemic clone and the accumulation of an increased number of defects in oncogene and tumor suppressor pathways.
Leukemia Cell Dissemination
The ability of the leukemic cell to egress from the marrow and invade tissues is poorly understood. t(8;21)-associated AML is associated with chloromas, and this phenotype is recapitulated in the murine model.12 Whether this is due to a unique pattern of expression of cell surface proteins programmed by the RUNX1 fusion protein is unknown. High level of selectin expression on the surface of leukemic cells is a negative prognostic marker in AML. Secretion of tumor necrosis factors and other cytokines by the leukemic blast can lead to increased expression of selectins, cadherins and other adhesion proteins on vascular endothelium, resulting in increased leukemia cell adhesion.40 Antibodies directed against these surface proteins block adhesion. It is interesting to speculate that monoclonal antibodies directed against cell surface proteins on the leukemic cell such as anti-CD33 could work in part by interfering with adhesion interactions important for stimulation of cell growth or cell migration.
Conclusion
The molecular pathogenesis of AML is complex, but the multiple genetic defects that have been described can be understood to converge on a set of biological properties of the leukemic cell (Figure 1; see Color Figures, page 545). In some cases there is a direct correlation between the molecular defect and the biological process, while in other cases there may be an indirect or an as yet undetermined explanation for the molecular basis of the biology of the disease. Through a more complete understanding of these links we anticipate that more precise and specific therapies for AML can be developed. Whereas the differentiation block and proliferative activity of AML is being targeted by current investigational therapies, methods to restore genomic stability, induce specific apoptosis of leukemia cells and restore cell cycle checkpoint control should be given strong consideration in the development of future experimental therapeutic efforts.
The molecular lesions in acute myeloid leukemia (AML) associated with malignant characteristics.
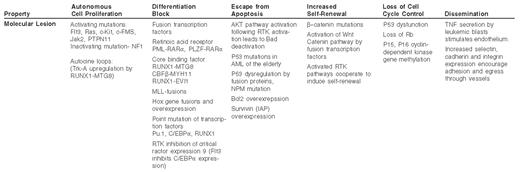
Abbreviations: TNF, tumor necrosis factor; AML, acute myelogenous leukemia; RTK, receptor tyrosine kinase
Supported by NIH RO1 CA59936 and a Burroughs Wellcome Clinical Scientist Award in Translational Research. DWS is supported by NIH K08 CA82261.
