
Abstract
Intensive chemotherapy regimens for children with acute lymphoblastic leukemia (ALL) have greatly improved, and the majority of children with precursor B-cell ALL are able to achieve a complete remission (CR), with an induction rate approaching 98% and a 5-year estimated event-free survival rate (EFS) of approximately 80%. Although there have been dramatic improvements over the last several decades in both the EFS and overall survival (OS) rates in young children with ALL, the results in adult clinical trials have not kept pace. Current adult treatment regimens result in CR rates in the 80% range, with EFS at 5 years of only 30%–40%. Adolescents and young adults represent a minority of patients enrolled onto either adult or pediatric clinical trials. As a result, little information is available regarding CR, EFS, and OS rates for this age group, and the appropriate treatment regimen for this group of patients remains elusive. Recent studies suggest that young adult patients have far superior outcomes when treated on more intensive pediatric regimens. In addition, new insights into the molecular pathogenesis of T cell ALL have led to new therapeutic strategies.
Treatment of Adults with Acute Lymphoblastic Leukemia
The overall results of adult clinical trials for patients with acute lymphoblastic leukemia (ALL) have been less encouraging than those trials conducted in pediatric age groups.1–5 The Cancer and Leukemia Group B (CALGB) initiated a five-drug induction regimen (protocol 8811) that added cyclophosphamide to their prior studies6 and adopted the consolidation phase from the German BFM (Berlin-Frankfurt-Munster) multicenter trials. This trial also made early and extensive use of l-asparaginase. With a median age of 32 years, the complete remission (CR) rate was 86% with a 3-year disease-free survival (DFS) of 46% and a 3-year overall survival (OS) of 50%. A subsequent CALGB study (protocol 9111) that randomized patients to the addition of filgrastim (G-CSF) resulted in a statistically significant improvement in CR rate, at 87% with G-CSF versus 77% with placebo; however, no improvements were seen in the 3-year DFS rate of 41% or OS rate of 43%.7 In a recent trial using the A-B-C regimen (protocol 19802), which is characterized by dose-intensive daunorubicin induction followed by high-dose methotrexate and cytarabine consolidation cycles, the CR rate was only 78% with a median OS of 19 months.8 In part the declining OS on these trials may be a representation of the older median age of 40 years, compared to a median age of 32 years for patients on CALGB 8811. Younger patients enrolled on these trials did have a markedly improved outcome compared to older patients. For example, patients under 30 years of age treated on CALGB 8811 had a CR rate of 94% with a DFS rate of 51% and an OS rate of 69%, as compared to a CR rate of 85% for patients between the age of 30 and 59 with DFS and OS rates of only 43% and 39%, respectively.
The CALGB studies also demonstrated several important points. First, the use of l-asparaginase is well tolerated in adult patients. Second, patients with T cell ALL (T-ALL) actually fared better than patients with pre-B cell ALL, especially if they presented with a mediastinal mass. Although the CR rates for patients with T-ALL and pre-B cell ALL were similar, the OS rate was 62% with T-ALL as compared to 38% with pre-B cell ALL.6 Third, the coexpression of aberrant myeloid antigens on either the pre-B cell or T cell immunophenotypes did not alter the overall prognosis.
Many other adult ALL regimens exist, but all have different age and study entry criteria, thereby making direct comparisons difficult. Kantarjian and colleagues at MD Anderson Cancer Center have reported results using their hyper-CVAD regimen,9,10 which consists of alternating cycles of cyclophosphamide, vincristine, doxorubicin, and dexamethasone with cycles of methotrexate and cytarabine. With a median age of 40 years, 92% of the patients achieved a CR and the 5-year DFS and OS rates were both 38%. Again, younger patients outperformed their older counterparts. For example patients under 30 years of age achieved a CR rate of 98% and an OS rate of 54%, as compared to CR and OS rates of 89% and 42% for patients between the ages of 30 and 49. The French LALA-94 multicenter trial demonstrated a CR rate of 84% and a 3-year EFS rate of only 37%. Similar results have been reported from the Medical Research Council (MRC) and the German multicenter trials.11–13 Clearly the CALGB, MD Anderson, and the French, German, and MRC multicenter trials have made important improvements in the outcome of adult ALL. However, the question of whether all adult patients, regardless of age, should be treated in a similar fashion remains to be answered.
Each of the adult clinical ALL trials described above included patients over age 15 and extended up to age 80 years on the CALGB trials and age 79 on the hyper-CVAD regimen. Although there was only a small decrement in CR rates with advancing age, the OS rates were far superior in younger patients as compared to those of older patients treated with the same regimen. Despite the better results seen in younger patients treated on adult trials, it is possible that younger individuals treated with these regimens might still be receiving less intensive and therefore inferior therapy.
An additional consideration is that the role of each individual component has not been well studied within the various adult ALL regimens. Pediatric ALL clinical trials have clearly shown that dexamethasone is superior to prednisone in optimizing the rate of DFS (85% vs 77%; P = 0.002), mainly by reducing the risk of central nervous system (CNS) relapse.14,15 Furthermore, dose intensification of the non-myelosuppressive chemotherapy agents, such as vincristine, corticosteroids, and asparaginase, has improved the OS rates in young children on many protocols.4,16
Treatment of Adolescents and Young Adults with Acute Lymphoblastic Leukemia
Clear biologic differences emerge with the advancing age of patients with ALL. These are reflected in the age-dependent outcomes for patients with pre-B cell ALL (Table 1 ). The most favorable outcome is achieved in children aged 1 to 5 years old, in which the EFS rate is greater than 80%.16,17 The rate decreases to approximately 60% in adolescents aged 15 to 18 years old and decreases further to less than 40% in adults.6,7,9,11–13 By comparison, T cell ALL, which accounts for about 25% of the cases of ALL above age 10, is associated with an average EFS rate of 60%, independent of age.6,18,19
The most important predictor of outcome in patients with ALL is the acquired genetic and molecular characteristics of the lymphoblasts. Cytogenetic abnormalities consist of either a chromosomal translocation or altered number of chromosomes (Table 2 ). The most important cytogenetic abnormality is the Philadelphia chromosome [Ph+; t(9;22)(q34;q11)]. In studies using conventional chemotherapy, Ph+ ALL was associated with an OS rate of less than 10% when stem cell transplantation was not included in the treatment regimen.20 Although higher remission rates have been reported with combination chemotherapy and imatinib, it is still unclear whether or not this will result in better long term survival data. The frequency of Ph+ ALL is remarkably age dependent. It is found in less than 3% of children under age 18 years, up to 6% of all patients under age 25, but as high as 14% among individuals between 25 to 35 years of age, 33% among patients 36 to 55 years of age, and up to 53% among patients older than age 55 years.21
The single most common cytogenetic abnormality is hyperdiploidy (≥ 47 chromosomes), which is found in almost one third of all cases of childhood pre-B cell ALL, but in less than 6% of adult cases.22 Children with a “high” hyperdiploid state (chromosome number between 51 and 63) have an extremely low risk of relapse, with EFS rate of 75%–90%. In contrast, hypodiploidy (< 46 chromosomes) is found in approximately 5% of both adult and pediatric cases of ALL and is associated with a poor outcome.23 Another age-dependent cytogenetic aberration is t(12;21) or the TEL-AML1 molecular counterpart. This abnormality, identified by using molecular screening techniques, is found in about 25% of children, but is rare in patients older than 18 years of age.24,25 Patients with the TEL-AML1 translocation have an extremely high OS rate, approaching 90%, which may be due to the relative sensitivity of the TEL-AML1 lymphoblasts to chemotherapy.26 Thus, as the median age increases in ALL, there is an overall increase in the frequency of adverse cytogenetic features and a remarkable decrease of the favorable abnormalities (Table 3 ).
Since age is a continuous variable, setting discrete limits for classification purposes and clinical trial participation is rather arbitrary. Currently, an 18-year-old patient referred to a pediatric hematologist is treated rather differently from the same patient who happens to be referred to an adult oncologist. Recent data suggest that the outcome of older adolescents (aged 15–20 years) with ALL is markedly improved if they are treated on intensive pediatric protocols rather than on less intense adult ALL protocols. A retrospective study from France compared patients aged 15–20 years that were treated either on the pediatric FRALLE-93 protocol or on the adult LALA-94 protocol.27 The CR rate for the 77 patients treated on the pediatric trial was 94% compared to 83% for the 100 patients treated on the adult LALA-94 trial. With a median follow-up of 3.5 years, the estimated 5-year EFS and DFS rates for the patients who achieved a CR were superior for those treated on the pediatric as opposed to the adult clinical trials (67% vs 41% and 72% vs 49%) (Table 4 ). Although the median age of patients in the LALA-94 study was 2 years older than those in the pediatric trial, the cohorts were well matched for sex, immunophenotype, and cytogenetic profiling, and a multivariate analysis revealed an independent influence of the treatment regimen on prognosis. Differences in drug and dose intensity may explain the superior results with the pediatric FRALLE-93 regimen. That study used five times more prednisone than the adult LALA study. Also, 50% more prednisone was used during the induction course, and three times more vinca alkaloids and 20 more doses of l-asparaginase were administered in the FRALLE study as compared to the LALA-94 trial. In the adult trial, the use of vinca alkaloids was restricted to the induction and consolidation courses, and asparaginase was not administered during the induction phase of the trial. Interestingly, adherence to both trials was similar; however, the time between achieving CR and the administration of the first post-remission dose of chemotherapy was only 2 days in the FRALLE study as compared to 7 days in the LALA study (P = 0.0002), and only 15% of patients treated on the FRALLE study had an interval of 7 or more days.
The CALGB and the Children’s Cancer Group (CCG) have presented a similar analysis.28 They compared patients aged 16 to 21 years who were treated between 1988 and 1998 on sequential trials. A large retrospective cohort analysis compared 103 patients treated on CALGB studies to 196 patients treated by various CCG centers. The groups were surprisingly well controlled for age, sex, immunophenotype, white blood cell (WBC) count, and cytogenetic abnormalities. Although the CR rates were similar between the groups treated by the CCG (96%) and CALGB (93%), the 6-year EFS rates were highly discordant, with 64% among those treated on CCG trials and only 38% among similar patients treated by the CALGB (Table 4 , Figure 1 ).
Investigators from the Netherlands reported similar results for patients aged 15–21 years.29 The 5-year DFS rate was 69% for those treated on the pediatric DCOG protocol compared to only 34% for patients treated on the adult HOVON protocols (ALL-5 and ALL-18). In these studies, the more favorable outcome for older adolescents on pediatric protocols was not explained by differences in patient characteristics, but solely on whether they were treated on the more intensive pediatric regimen. Italian studies also showed an inferior outcome when patients aged 14 to 18 years were treated on adult rather than the more intensive pediatric regimens (Table 4 ).30 By contrast, the MRC used identical treatment regimens (UKALL X and XA) for both pediatric and adult patients and found dramatic differences in both the DFS as well as the OS, which has led some investigators to suggest that pediatric regimens may not be optimal for adult patients.5
Why adolescent adults fare more favorably when treated on pediatric ALL protocols is unclear.31 Dose intensity is clearly greater within the pediatric protocols, especially with such agents as asparaginase, vincristine, corticosteroids, and methotrexate. For example, the dose of vincristine was capped at 2 mg in the adult trials. However, the CALGB regimen was inferior to the CCG protocol despite the administration of 14 doses of asparaginase over a 7-week period within the first 3 months. The choice of antileukemic agents may also play a role. Although cyclophosphamide was used in the CALGB, hyper-CVAD, and LALA trials, and not in the FRALLE study, it did not appear to offer benefit, as predicted from an earlier study.32
Differences in therapeutic practices may be equally important. Frequently, longer delays are encountered during therapy on adult trials as compared with pediatric protocols. In addition, pediatric hematologists typically administer chemotherapy agents with greater adherence to schedules and dose density. Another disturbing realization is that where a patient undergoes treatment, especially a young adult, can translate into a dramatic difference in survival. In the US and Canada, patients between 16 and 21 years of age with ALL or AML had a superior outcome if they were treated on CCG trials as compared to those patients who were not entered.18 Although many confounding variables exist, this observation may apply to other diseases as well. Young adults with Ewing’s sarcoma had a better outcome when they were treated at pediatric centers rather than adult centers, even when they got the same drug regimen.33 This has led some physicians to suggest that psychosocial factors must be considered in addition to biology. No published studies are yet available in which both children and adults with ALL have been treated on the same treatment protocol with uniform therapy.
A reasonable strategy for pediatric and adult patients younger than middle age is to develop and implement age-unrestricted but disease-specific treatment protocols. With this approach it can be hoped that the striking improvement in the success of therapy of childhood ALL can be translated to improved survival in adults. A further consideration is that young adults might best be treated at pediatric centers where they would be more likely to benefit from what is referred to as the “mother factor,” i.e., a situation in which a patient is more compliant because their care is directed by their parents. One must remember that ALL remains the most common leukemia found in children but is much less common in adults. Therefore, pediatric centers may simply be better at implementing chemotherapy regimens targeted at pediatric cancers, even when using the same protocols for both children and young adults. Within this context, intensified chemotherapy regimens might be more accurately assessed, as exemplified by the Pediatric Oncology Group experience in which a randomized study demonstrated improved outcome for children with T-cell ALL,34 a subtype that is more common in adolescents and adult patients. To minimize toxicity without compromising efficacy, it is important to optimize the dosing of currently available chemotherapeutic agents, such as asparaginase, doxorubicin, vincristine, and corticosteroids, in adolescents and young adults. It is also essential that new agents be identified to treat this disease more effectively and with less toxicity.
Clinical Trial Participation of Adolescents with Cancer
Both pediatric and adult oncologists, depending on the local referral patterns, treat patients between 16 and 25 years. In this age group, cancer remains a leading disease cause of death, fourth behind accidents, suicides, and homicides. Unfortunately, only about 5% of all 15- to 25-year-olds with cancer are entered onto US clinical trials compared to 60%–65% of younger children (Figure 2 ).35,36 This low accrual occurs despite the fact that cancer is diagnosed more commonly in this age group than in the 5- to 9-year and 10- to 14-year age groups. It should not be surprising, therefore, that the improvement in the cancer mortality rate among 15- to 29-year-olds has lagged behind the dramatic improvements in mortality reduction seen in younger patients (Figure 3 ). The incidence of ALL declines steadily with age. It accounts for 30% of all cancers in children less than 15 years but only 6% in adolescents. The declining incidence of ALL in adolescents and young adults may be the principal reason behind the deficit in specific treatment regimens for this age group. French, US, Dutch and Italian studies suggest that development of such regimens can dramatically affect OS rates.
The lack of clinical trial participation and initiatives for age-specific treatment protocols may be due to the underutilization of the available health care services by this age group as well as the fact that young adults in the US are typically under-insured or carry no health insurance benefits, further compromising their access to health care. In addition, comprehensive cancer centers are more dedicated to the treatment of cancers of either younger or older patients, leaving the adolescent and young adult patient somewhat disconnected. Clearly, the treatment of cancer during adolescence and young adult life remains a specific and unmet challenge.
Current Strategies in the Treatment of Younger Adults with ALL
At the Dana-Farber Cancer Institute (DFCI), we are currently testing an age-unrestricted approach to all patients with ALL. Historically, the outcome for the oldest evaluable patients treated in the DFCI Childhood ALL Consortium protocols, namely those aged 15–18 years, has been relatively favorable, with a 5-year EFS of 77% (52 patients treated between 1991 and 2000). This result compares favorably to the overall 5-year EFS rate of 83% for all children ages 1 to 18 years treated on these studies.4 Therefore, since late 2002, we have enrolled adults between the ages of 18 and 50 years onto a pilot protocol with therapy identical to the high-risk arm of the DFCI Consortium pediatric protocol (no. 00–01). The principal objective of this study is to determine the feasibility and toxicity associated with a dose-intensive pediatric regimen in adults with newly diagnosed ALL.
As of July 2005, 34 eligible adult patients have been enrolled and have completed remission induction therapy. The mean age is 31 years (range 19–48 years). Thirty-three percent had a T-cell phenotype and 18% Ph+ disease. Thus far, 79% (27 of 34) have achieved complete remission, including 83% of patients with Ph+ disease without the addition of imatinib. Six patients had persistent leukemia at the end of remission induction, including one patient with Ph+ disease. The remaining four Ph+ patients were taken to stem cell transplant after remission induction, as per protocol. Of the 28 non-Ph+ patients, 18 have completed post-remission intensification, and 14 received 26 or more of the 30 intended doses of asparaginase. Only 1 patient out of the remaining non-Ph+ patients has suffered a relapse. Although the median follow-up is short, this experience suggests that dose-intensification of young adult patients with newly diagnosed ALL is possible without significantly increasing treatment toxicity. A new trial will be launched this year by CALGB and COG using the COG AALL0232 high-risk pediatric regimen to examine survival rates of patients with ALL at 15–30 years of age and compare these with patients in other age groups and with patients in different age groups treated in previous studies.
Pathogenesis of T-Cell Acute Lymphoblastic Leukemia
T-ALL is observed primarily in older children, adolescents, and young adults (Table 3 ). Although a more favorable de novo phenotype than pre-B cell ALL in adults (Table 1 ), recurrent or refractory T-ALL is frequently resistant to standard chemotherapeutic agents, and sustained second or subsequent remissions occur in a very small minority of patients. Thus, novel and targeted approaches for T-ALL would benefit many patients. As demonstrated by the German multicenter trials as well as the DFCI and MD Anderson, the OS rates of patients with T-cell lymphoblastic lymphoma (T-LBL) are dramatically improved when these patients are treated using ALL regimens.37–39
Ferrando et al have identified five different T cell oncogenes (HOX11, TAL1, LYL1, LMO1, and LMO2) that are aberrantly expressed in T-ALL and are usually found in the absence of detectable chromosomal abnormalities.40 These five oncogenes are associated with gene expression signatures that are indicative of a developmental arrest at a specific stage of thymocyte development, and as such, they may provide important insights to pathogenesis and treatment. Regarding the latter, patients with a HOX11 mutation have an extremely favorable outcome, with an OS rate of greater than 90%. This phenotype suggesting high drug susceptibility may be due to overexpression of genes involved in proliferation and the lack of BCL2 expression.
The NOTCH1 signaling pathway has also recently been shown to be dysregulated in T-ALL. The mammalian NOTCH1 gene was originally identified because of its involvement in the t(7;9) chromosomal translocation, a cytogenetic abnormality that is only rarely detected in human T-ALL. Subsequently, NOTCH1 expression was shown to be essential for normal development of T cell progenitors (Figure 4 ).41,42 Gain-of-function mutations of NOTCH1 can reliably produce T-ALL in animal models. Recently published data demonstrated that lymphoblasts from 50%–60% of patients with T-ALL have NOTCH1 gain-of-function mutations, suggesting that NOTCH1 plays a critical role in the pathogenesis of T-ALL.43 The extent to which aberrant NOTCH1 signaling contributes to human T-cell ALL remains undefined.44
To generate critical downstream signals, most mutated forms of NOTCH1 require the activity of the gamma-secretase enzyme. Weng and colleagues demonstrated that gamma-secretase inhibitors completely abrogate the stimulatory effects of mutated transmembrane NOTCH1 polypeptides, and strongly inhibit the proliferation of NOTCH1-mutated human T-ALL cell lines.43 These findings provide a strong rationale to test gamma-secretase inhibitors in T-cell ALL. The Dana-Farber Cancer Institute and the University of Chicago are currently enrolling patients with relapsed or refractory T-ALL or T-cell lymphoblastic lymphoma onto a Phase I/II trial of the Notchsecretase inhibitor, MRK002, which was developed by Merck.
Other novel genetic mutations have been recently reported in T-ALL. The NUP214-ABL1 fusion is a constitutively activated tyrosine kinase that transforms Ba/F3 cells to factor independent growth. In a recently published study, leukemic cells from 5 of 65 patients demonstrated the NUP214-ABL1 fusion solely contained within episomal DNA.44 The NUP214-ABL1 fusion appears to be present in ~5%–10% of patients with T-ALL and represents at least one example of a cryptic mutation that results in the activation of a tyrosine kinase in a significant fraction of patients with T-ALL. Importantly, the NUP214-ABL1 fusion gene product is inhibited by imatinib, thereby suggesting its use as a therapeutic strategy in relapsed T-ALL with NUP214-ABL1 fusion.
New Agents for the Treatment of T-ALL
A major obstacle to the introduction of new agents in ALL has been, ironically, the success of currently available cytotoxic therapies for disease in children. Even at the time of first relapse, durable remissions can be achieved with conventional chemotherapy and/or stem cell transplantation, and so investigators have been reluctant to risk compromising therapy with newer agents whose efficacy and toxicity profiles are less well known. With relatively few patients available in second or later relapse, it is challenging to gauge the efficacy of a promising new drug. Furthermore, once safety and efficacy have been confirmed, it is even more challenging to demonstrate that outcome can be improved in a disease that already has an 80% cure rate. For these reasons, large phase II and III trials can and should be conducted within the national cooperative group setting, in which greater numbers of patients can allow for definitive efficacy assessments of new agents.
Nelarabine (compound 506U78) has substantial clinical activity in T-ALL.45,46 Preclinical studies demonstrated that T-ALL blasts are extremely sensitive to the cytotoxic effects of deoxyguanosine and its analog AraG. AraG is poorly water soluble, but nelarabine is a soluble pro-drug. Nelarabine is rapidly demethylated in the serum by adenosine deaminase to AraG. As predicted by the preclinical studies, the highest response rates in early phase I studies were observed in patients with T-cell ALL as compared to patients with other T-cell malignancies.47 Berg et al recently reported results from a trial conducted by the Children’s Oncology Group (COG) in children with refractory T-cell malignancies treated for 5 consecutive days.48 An overall response (OR) (CR plus partial response [PR]) rate of 55% was seen in patients with T-ALL in first relapse and an OR rate of 27% was seen with T-ALL in second relapse.49 Only 14% of patients with T-cell lymphoma responded to nelarabine. Grade 3 or 4 neurologic adverse events were seen in 18% of the patients, including peripheral neuropathy, hallucinations, seizures, and one episode of a Guillain-Barré–like syndrome. The CALGB conducted a similar study in patients with multiply relapsed T-ALL and T-LBL using a dose of 1.5 g/m2 on days 1, 3, and 5.49 An extremely low rate of neurologic toxicity was seen with this treatment regimen. Of the 21 evaluable patients with T-ALL, 6 achieved a CR and 2 a PR for a total response rate of 38%. For the 17 evaluable patients with T-LBL, 4 achieved a CR and none a PR, for a total response rate of 24%. The OR rate for all of the 38 evaluable patients was 32%. These data suggest that nelarabine has significant activity in T-ALL, especially for those patients who are in first relapse, and that studies using nelarabine in patients with newly diagnosed T-ALL are warranted.
Conclusions
With the remarkable success in the treatment of childhood ALL, it is now time to reexamine the treatment of adolescent and young adult patients. It is likely that adolescents and young adults are currently being underdosed with the standard adult ALL regimens. The tolerability and efficacy of regimens that treat pediatric and younger adult patients with dose-intensive chemotherapy should be investigated. The new CALGB/COG study will test this hypothesis in patients less than 30 years, while we at the Dana-Farber are currently treating patients ages 1 to 50 years using the same treatment protocol. The importance of referring patients with ALL to major academic treatment centers with established expertise cannot be overemphasized. The vast majority of children with ALL are referred to large academic centers and treated on clinical trials by physicians who focus on ALL, whereas the majority of adults are being treated by adult oncologists. Those adult patients are not being enrolled onto clinical trials nor, for the most part, are they being treated by experienced support staff. A major goal is to increase participation of adolescents and young adults in clinical trials that address specific treatment issues, with the hope that novel approaches will lead to improved survival rates for patients of all ages with ALL.
Immunophenotypes and survival in acute lymphoblastic leukemia (ALL) in children (ages 1–18) versus adults (ages 18 and older).
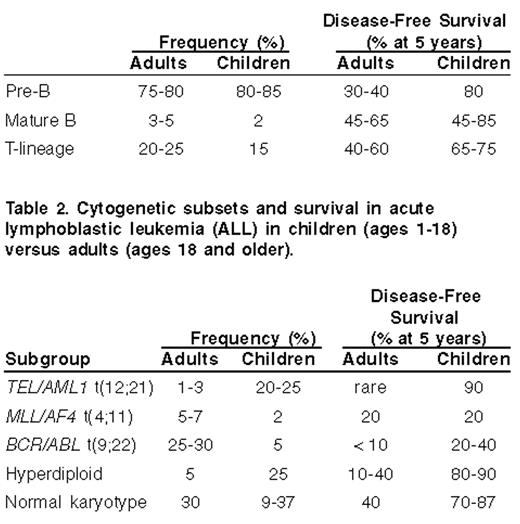
Cytogenetic subsets and survival in acute lymphoblastic leukemia (ALL) in children (ages 1–18) versus adults (ages 18 and older).
The association of age on immunophenotype and cytogenetics in acute lymphoblastic leukemia (ALL).
| Age (years) | |||||
|---|---|---|---|---|---|
| Subgroup | 1–9 | 10–14 | 15–19 | 20–39 | 40+ |
| This data was extracted from the UKALL X and XA studies5 with the exception of the TEL-AML1 data, which was extracted from the ALL-AIEOP95 and ALL-BFM95 trials.25 | |||||
| B-cell | 86 | 68 | 70 | 60 | 75 |
| T-cell | 6 | 22 | 19 | 20 | 8 |
| Ploidy: | |||||
| Normal | 39 | 44 | 30 | 37 | 34 |
| Hypodiploid | 5 | 8 | 7 | 6 | 7 |
| Hyperdiploid | 37 | 20 | 29 | 16 | 15 |
| Tri-tetraploid | 2 | 3 | 4 | 1 | 8 |
| Chromosomes: | |||||
| TEL/AML1 t(12;21) | 24 | 18 | 5 | 0 | — |
| PBX1/E2A t(1;19) | 2 | 3 | 2 | 3 | 4 |
| MLL/AF4 t(4;11) | 1 | 2 | 2 | 0 | 9 |
| BCR/ABL t(9;22) | 1 | 3 | 4 | 12 | 19 |
| Age (years) | |||||
|---|---|---|---|---|---|
| Subgroup | 1–9 | 10–14 | 15–19 | 20–39 | 40+ |
| This data was extracted from the UKALL X and XA studies5 with the exception of the TEL-AML1 data, which was extracted from the ALL-AIEOP95 and ALL-BFM95 trials.25 | |||||
| B-cell | 86 | 68 | 70 | 60 | 75 |
| T-cell | 6 | 22 | 19 | 20 | 8 |
| Ploidy: | |||||
| Normal | 39 | 44 | 30 | 37 | 34 |
| Hypodiploid | 5 | 8 | 7 | 6 | 7 |
| Hyperdiploid | 37 | 20 | 29 | 16 | 15 |
| Tri-tetraploid | 2 | 3 | 4 | 1 | 8 |
| Chromosomes: | |||||
| TEL/AML1 t(12;21) | 24 | 18 | 5 | 0 | — |
| PBX1/E2A t(1;19) | 2 | 3 | 2 | 3 | 4 |
| MLL/AF4 t(4;11) | 1 | 2 | 2 | 0 | 9 |
| BCR/ABL t(9;22) | 1 | 3 | 4 | 12 | 19 |
| Cooperative Group/Study Period | No. of Patients | CR (%),5-year | EFS (%),5-year |
|---|---|---|---|
| * 6-year EFS | |||
| † DFS | |||
| Abbreviations: CR, complete remission; EFS, event-free survival; DFS, disease-free survival | |||
| North America 1988–1998 Age of patients (yrs): 16–21 | |||
| CCG 1882 (peds) | 196 | 96 | 64* |
| CALGB 8811-9511 (adult) | 103 | 93 | 38* |
| French 1993–1994 Age of patients (yrs): 15–20 | |||
| FRALLE-93 (peds) | 77 | 94 | 67 |
| LALA-94 (adult) | 100 | 83 | 41 |
| Dutch 1985–1999 Age of patients (yrs): 15–20 | |||
| DCOG-ALL (peds) | |||
| 15–18 yrs | 47 | 98 | 69 |
| HOVON (adult) | |||
| 15–18 yrs | 44 | 91 | 34 |
| 19–20 yrs | 29 | 90 | 34 |
| Italian 1996–2000, Age of patients (yrs): 14–18 | |||
| AIEOP (peds) | 153 | 94 | 83† |
| GIMEMA (adults) | 95 | 95 | 55† |
| Cooperative Group/Study Period | No. of Patients | CR (%),5-year | EFS (%),5-year |
|---|---|---|---|
| * 6-year EFS | |||
| † DFS | |||
| Abbreviations: CR, complete remission; EFS, event-free survival; DFS, disease-free survival | |||
| North America 1988–1998 Age of patients (yrs): 16–21 | |||
| CCG 1882 (peds) | 196 | 96 | 64* |
| CALGB 8811-9511 (adult) | 103 | 93 | 38* |
| French 1993–1994 Age of patients (yrs): 15–20 | |||
| FRALLE-93 (peds) | 77 | 94 | 67 |
| LALA-94 (adult) | 100 | 83 | 41 |
| Dutch 1985–1999 Age of patients (yrs): 15–20 | |||
| DCOG-ALL (peds) | |||
| 15–18 yrs | 47 | 98 | 69 |
| HOVON (adult) | |||
| 15–18 yrs | 44 | 91 | 34 |
| 19–20 yrs | 29 | 90 | 34 |
| Italian 1996–2000, Age of patients (yrs): 14–18 | |||
| AIEOP (peds) | 153 | 94 | 83† |
| GIMEMA (adults) | 95 | 95 | 55† |

The event-free survival (EFS) of young adults ages 16–21 with acute lymphoblastic leukemia (ALL) treated on CCG and CALGB trials from 1988–1995.
The event-free survival (EFS) of young adults ages 16–21 with acute lymphoblastic leukemia (ALL) treated on CCG and CALGB trials from 1988–1995.


A. The number of patients entered onto US clinical trials for patients less that 45 years of age between 1997 and 2003; B. the number of patients less than 45 years of age entered onto US clinical trials for patients with leukemia between 1997 and 2003.
A. The number of patients entered onto US clinical trials for patients less that 45 years of age between 1997 and 2003; B. the number of patients less than 45 years of age entered onto US clinical trials for patients with leukemia between 1997 and 2003.

The comparison of the average percent change in 5-year survival from 1975 to 1997 based on US SEER data.
The comparison of the average percent change in 5-year survival from 1975 to 1997 based on US SEER data.

NOTCH1signaling governs lymphoid cell fate decisions lymphogenesis.
Abbreviations: CLP, common lymphoid progenitor; BM, bone marrow; ICN1, intracellular Notch1
NOTCH1signaling governs lymphoid cell fate decisions lymphogenesis.
Abbreviations: CLP, common lymphoid progenitor; BM, bone marrow; ICN1, intracellular Notch1
