
Abstract
In this chapter, Drs. Keating and Willman review recent advances in our understanding of the pathophysiology of acute myeloid leukemia (AML) and allied conditions, including the advanced myelodysplastic syndromes (MDS), while Drs. Goldstone, Avivi, Giles, and Kantarjian focus on therapeutic data with an emphasis on current patient care and future research studies.
In Section I, Dr. Armand Keating reviews the role of the hematopoietic microenvironment in the initiation and progression of leukemia. He also discusses recent data on the stromal, or nonhematopoietic, marrow mesenchymal cell population and its possible role in AML.
In Section II, Drs. Anthony Goldstone and Irit Avivi review the current role of stem cell transplantation as therapy for AML and MDS. They focus on data generated on recent Medical Research Council studies and promising investigation approaches.
In Section III, Dr. Cheryl Willman reviews the current role of molecular genetics and gene expression analysis as tools to assist in AML disease classification systems, modeling of gene expression profiles associated with response or resistance to various interventions, and identifying novel therapeutic targets.
In Section IV, Drs. Hagop Kantarjian and Francis Giles review some promising agents and strategies under investigation in the therapy of AML and MDS with an emphasis on novel delivery systems for cytotoxic therapy and on targeted biologic agents.
I. Biology of Acute Myeloid Leukemia: The Role of Stroma
Armand Keating, MD*
Medical Oncology & Hematology, Princess Margaret Hospital, 610 University Avenue, Suite 5-211, Toronto, ON M5G 2M9, Canada
Over the past decade, rapid advances have been made in elucidating some of the key molecular lesions that lead to acute myeloid leukemia (AML).1 The therapeutic implications of a detailed knowledge of aberrant signal transduction in malignant cells are evident, as the success of imatinib mesylate in the treatment of chronic myeloid leukemia dramatically attests. It is perhaps not surprising, then, that considerably less attention has been directed toward examining the role of the hematopoietic microenvironment (HM) in the initiation and progression of leukemia. The definition of the HM as an entity that regulates hematopoiesis through interactions with progenitor cells, hematopoietic cytokines, and the biosynthetic products of stromal and other cells suggests, however, that much may be learned about the leukemic state by a better understanding of this area. The recent resurgence of interest in the stromal or nonhematopoietic marrow mesenchymal cell population may serve as a springboard for further studies of how stroma influences, or is influenced by, leukemia. A brief overview of the normal HM will serve as a prelude to a review of stromal cells in AML.
The Normal Hematopoietic Microenvironment
The HM in the bone marrow consists of a heterogeneous population of hematopoietic and nonhematopoietic stromal cells, their extracellular biosynthetic products, and hematopoietic cytokines (reviewed in Clark and Keating2). The cells include myofibroblasts, other fibroblastoid cells, endothelial cells, osteogenic precursors, adipocytes, and macrophages. These cells produce a complex array of extracellular matrix (ECM) molecules consisting of proteoglycans and their constituent sulfated glycosaminoglycans, chondroitin, heparan, and dermatan species as well as hyaluronic acid.2 In addition, they make a variety of interstitial (fibril-forming) and basal lamina collagens, including collagen types I, III, IV, V, and VI. Stromal cells also synthesize other matrix molecules, such as fibronectin, thrombospondin, hemonectin, sialoadhesin, laminin, and the tenascin glycoproteins (reviewed in Klein3 and Verfaillie et al4) (Table 1 ). Cells comprising the HM also provide a source of many hematopoietic cytokines, either secreted or membrane bound, including GM-CSF, G-CSF, and stem cell factor (kit ligand) (Table 2 ).
The growth, differentiation, and survival of hematopoietic stem/progenitor cells is regulated in the HM by at least three different mechanisms that involve the following:
Interactions of hematopoietic progenitor cells with hematopoietic cytokines, present in the HM, in part in association with ECM components such as glycosaminoglycans
Interactions between hematopoietic and stromal cells by means of cell adhesion molecules
Interactions of adhesion molecules on hematopoietic cells with appropriate ligands on ECM components
Evidence is emerging that, in addition to hematopoietic cytokines, adhesion molecules are involved in mediating signal transduction in hematopoietic precursors and hence play an important role in cell proliferation that extends beyond brokering hematopoietic cell contact and adhesion to stromal cells and ECM elements (reviewed in Levesque and Simmons5).
Interaction of Adhesion Molecules on Hematopoietic Precursors and Marrow Stroma
Integrins
The integrins are particularly important because they are involved in stromal cell–stem cell, as well as stem cell–ECM, adhesion. They are heterodimeric proteins consisting of noncovalently linked α and β chains that uniquely pair to form at least 20 different integrin receptors. The receptors are transmembrane structures in which the cytoplasmic domain initiates intracellular signaling involving the phosphorylation of cytoplasmic proteins and modulation of cell proliferation by activation of the ras and other pathways.5 The cytoplasmic portion of the integrin interacts with cytoskeletal elements (talin) that in turn influence the development of focal adhesion contacts between the cell and the ECM.
There are two main β integrin families involved in hematopoiesis, the β1 and β2 integrins. Binding specificity of the integrins is largely conferred by the particular α chain.
β1 Integrins
The β1 common chain (CD29) combines with different a chains to form a variety of VLA (very late antigen) molecules that mediate the adhesion of hematopoietic cells to ECM components and ligands on stromal and endothelial cells. CD34(+) cells express the integrin receptor VLA-4 (α4β1) (CD49d), whose ligand is VCAM-1 (vascular adhesion molecule-1) on marrow stromal cells and fibronectin in the ECM. VCAM-1 is variably expressed on marrow stromal and endothelial cells and can be upregulated by several cytokines, including interleukin-1 (IL-1). Because VLA-4 and its ligands are widely distributed, specificity is most likely conferred by the coexpression of other adhesion molecules and can be modulated by hematopoietic cytokines. The VLA-4/VCAM-1 interaction is a critical component of the complex process of stem cell homing.7 The chemokine and chemoattractant stromal-derived factor 1 (SDF-1), another important element in the homing of hematopoietic stem cells to the bone marrow, is secreted by stromal cells and strongly upregulates the VLA-4-mediated adhesion of CD34(+) cells to stroma and ECM fibronectin.8 Early precursors also express VLA-5 (α5β1) (CD49e), which can bind to ECM fibronectin.
β2 Integrins
Of the three β2 integrins (CD18), the best characterized is LFA-1 (lymphocyte function antigen-1) (CD11a), which is associated with the adhesion of more mature leukocytes to the ligand ICAM1 on endothelium but is also found on early hematopoietic precursors. Mac-1 (CD11b), another β2 integrin, is found on mature monocytic and granulocytic cells but not on early hematopoietic precursors.
Selectins
The selectins, a family of three glycoproteins, are also involved in adhesion and signaling. L-selectin is expressed not only on mature leukocytes but also on early hematopoietic precursors. Its role in adhesion is best documented in leukocyte attachment to endothelium. In contrast, P- and E-selectin are found on endothelial cells, while their ligands are found on early hematopoietic cells.
Sialomucins
Other adhesion molecules include the sialomucins, glycoproteins carrying O-linked sugars: CD34, CD45RA, leukosialin (CD43), and the more recently identified CD164 molecule (reviewed in Simmons et al9). CD164 (MGC-24) is expressed on both marrow stromal and CD34(+) cells and mediates adhesion between the two cell populations. Recent studies show that in addition, this molecule is involved in modulating the proliferation of early precursors.10 Other sialomucins, including leukosialin, also appear to act as negative regulators of hematopoiesis.11
Immunoglobulin superfamily
These cell adhesion molecules (CAMs) share a degree of sequence homology with immunoglobulins and are involved in cell-cell interactions. There are three main CAMs of relevance to hematopoiesis: VCAM-1, the ICAMs, and NCAM. VCAM-1 is expressed on marrow stromal cells and endothelial cells and interacts with the β1 integrin VLA-4 (α4β1) on early hematopoietic progenitors. Expression of VCAM-1 can be upregulated by a variety of cytokines, notably IL-1β and tumor necrosis factor-α (TNF-α). Stromal cells also express ICAM1, which can interact with the β2 integrins such as LFA-1 and Mac-1. NCAM-1 (CD56), which is a marker of natural killer (NK) cells and is found on neuronal tissue, is also expressed on marrow stromal cells and is involved in supporting lymphopoiesis.
CD44 proteoglycans
The CD44 family, highly expressed on stromal cells, binds the nonsulfated glycosaminoglycan hyaluronic acid, a major component of the ECM present in long-term marrow culture adherent layers. Although CD44 is also expressed on hematopoietic precursors, only a small proportion (presumably high-affinity receptors) binds hyaluronate.12 CD44 can also bind to other ECM components, including fibronectin.13 Because CD44 is widely expressed, specificity of interaction is conferred by numerous isoforms generated by alternative splicing. Anti-CD44 antibody-blocking studies suggest that CD44 interactions are important in maintaining hematopoiesis in long-term marrow cultures.14
Cadherins
The cadherins (E-, N-, and P-cadherin) are transmembrane glycoproteins that mediate calcium-dependent cell adhesion in embryonic development and in the maintenance of tissue architecture. Their role in hematopoiesis is unclear, although recent studies indicate that both E- and N-cadherin are expressed on stromal cells and a subset of CD34(+) cells and erythroid progenitors.15– 17 Their role in affecting leukemic cell development is unknown, but E-cadherin expression can be downregulated in AML blasts by hypermethylation mechanisms.
The Hematopoietic Microenvironment in AML
Given the multitude of interactions possible between hematopoietic progenitor cells and the HM, the acquisition of a leukemic clone may have numerous effects on this relationship and influence the clinical characteristics of the leukemia. At least three possible consequences to changes in leukemia cell–HM interactions have been proposed,11 as shown in Table 3 .
Adhesion Molecule Expression on Leukemic Cells
AML blasts express many of the adhesion molecules identified on normal hematopoietic precursors. Although differential expression has been documented, results have been variable, perhaps reflecting the heterogeneity of AML as defined by morphology. For example, while AML blasts from one subset of patients express the integrins VLA-1, -2, -3, and -6, not usually found on normal CD34(+) cells,18 results of another study show reduced VLA-2, -3, and L-selectin levels and increased VLA-5 expression19 or, indeed, the absence of transcripts for the α2, α3, or α6 chains of the β1 integrins in the blasts of yet other AML patients.20 Perhaps the most interesting observation is from Lyon, where a correlation was shown between the expression of VLA-4 on leukemic blasts and a high initial white count as well as extensive marrow involvement.21
Studies by the Westmead group in Sydney confirm that adhesion of AML blasts, at least in part, is mediated by the interaction of VLA-5 with ECM fibronectin as well as via both β1 (VLA-4) and β2 (LFA-1) integrin interactions with stromal cells.22–,24 Bendall and colleagues have also shown that the adhesion of AML blasts to marrow fibroblasts can be modulated by a variety of mechanisms, including the upregulation of stromal VCAM-1 by TNF-α and interferon γ.25
The adhesion of AML cells to ECM elements may explain the tenacity with which residual leukemic blasts may persist in the marrow. The blasts from all patients in a small cohort with AML expressed the sialylated Lewis x antigen, a ligand for E-selectin, on endothelial cells, suggesting a mechanism for migration across the vascular wall and into extravascular tissue.26
Further studies are required to determine whether differences in the expression of adhesion molecules on leukemic blasts influence cell trafficking and the clinical phenotype in AML, as appears to be the case for chronic myeloid leukemia.27
Stromal Interaction with Leukemic Cells
Leukemic cells, like their normal hematopoietic counterparts, are subject to the influence of the HM. Encounters between the HM and leukemic cells can affect the apoptosis, differentiation, and proliferation of AML blasts.
Cell-Cell Interactions
Direct contact of leukemic cells with stromal layers strongly inhibits the apoptosis of the leukemic cells.28 The reduction in apoptosis correlates with enhanced growth of clonogenic leukemic cells. The growth factors, stem cell factor (SCF), GM-CSF, and TNF-α in serum-free medium could achieve the same degree of inhibition of apoptosis in only half the cases studied. The co-culture of primary untreated AML cells with a stromal cell line in the presence of chemotherapy agents inhibits drug-induced apoptosis and increases the viability of leukemic clonogenic cells.29 The interaction of acute lymphoblastic leukemia (ALL) cells with stromal cells in the presence of chemotherapy drugs reduces the level of caspase 3 in leukemic cells and may account for the reduced apoptosis.30 These studies indicate that direct contact between leukemic and stromal cells enhances the survival of clonogenic leukemic cells and may explain how the small numbers of malignant cells remaining after chemotherapy are protected in vivo.
These observations contrast with the demonstration of the relative inhospitability of long-term marrow cultures to CML and AML progenitors, which are preferentially lost, allowing normal precursors to be reexpressed,31,32 and formed the basis for the clinical purging of grafts for patients undergoing intensive therapy and autotransplant.33,34 It is possible that, over the extended period of culture, the system does not provide critical survival factors for neoplastic clones. Loss of malignant progenitors is much less likely in the cultures of patients with advanced disease,35 suggesting that such limitations to growth in vitro are overcome and that normal hematopoiesis remains severely suppressed, if it exists at all.
Cytokines
The growth-promoting effects on leukemic blasts of cytokines such as G-CSF, SCF, GM-CSF, macrophage colony-stimulating factor (M-CSF), and IL-6 secreted by stromal cells have been documented.11 There is evidence that the secretion of IL-1β by leukemic cells can stimulate the release of G-CSF and GM-CSF from endothelial cells, which, in turn, may affect the proliferation of leukemic blasts.36 Hepatocyte growth factor (HGF) or scatter factor, a pleiotropic cytokine involved in hepatocyte morphogenesis, is secreted by marrow stromal cells and, in conjunction with other growth factors (GM-CSF, IL-3), can augment the growth of committed progenitors through interaction with its receptor, c-met, found on CD34(+) cells.37,38 Alone, HGF appears to selectively stimulate AML blast colony growth and promotes migration of leukemic cells.39
In addition to cytokines, stromal cells release other factors that may influence the behavior of leukemic cells. For example, SDF-1, a chemokine and chemoattractant involved in the homing of stem cells, may affect leukemic cell trafficking.40 SDF-1 appears to selectively attract FAB M4/5 AML blasts that express the chemokine receptor CXCR4, a mechanism that may, in part, explain the marrow and tissue infiltration in this AML subtype.41
Changes in Cellular Composition of the HM
Studies comparing bone marrow biopsies obtained from AML patients with those from normal donors reveal normal numbers of macrophages but increased numbers of alkaline phosphatase–positive stromal cells and endothelial cells.42 Increased microvessel density in untreated AML correlates with vascular endothelial growth factor (VEGF) and VEGF receptor levels on AML blasts. The frequency of endothelial cells normalizes when remission is achieved,43,44 suggesting that AML blasts may stimulate endothelial cell growth in a paracrine fashion and change the HM in AML. AML blasts also secrete platelet-derived growth factor (PDGF),45 a potent mitogen for marrow stromal cells,46 possibly accounting for the increased frequency of stromal cells in active AML. An alternative explanation is that marrow adipocytes transdifferentiate to fibroblastic cells in response to other signals.11,47
A more detailed understanding of the composition and characteristics of the AML stromal population has been provided by studying the long-term marrow culture adherent layer, arguably the best in vitro model of the HM, than has been possible by conducting investigations in vivo. Stromal layers generated from patients with AML frequently appear abnormal because the adherent population either is sparse or absent, or appears morphologically disorganized. Several studies show that fibroblast progenitors (CFU-F) are reduced in frequency in the majority of patients with AML48 but can be restored to normal levels during remission.49 The reduction in CFU-F levels is probably not due to dilution by high numbers of leukemic blasts. Macrophages and adipocytes can also be reduced in AML adherent layers.50 Although the elaboration of inhibitors of stromal progenitors by AML blasts has been proposed as an explanation,51 mechanisms are needed to reconcile the observation of increased stromal cells in biopsy specimens and the report that CFU-F levels can be increased during leukemia relapse.52 At least one consequence of reduced cellularity in the stromal population has been documented. Mayani and colleagues showed that for some patients with AML, the reduced frequency of CFU-F correlated with reduced M-CSF levels in the adherent layer conditioned medium.50 Of interest, AML layers with normal cellularity had normal levels of CFU-F and M-CSF.
A Functional Defect in AML Stromal Layers
The inability of AML stromal layers, including those with normal cellularity, to adequately support normal hematopoiesis is well documented.50 However, fibroblastic cells derived from AML cultures free of macrophages have a normal capacity to support committed progenitor growth.53 Suppression of hematopoiesis is attributed, in part, to the production of TNF-α and possibly, prostaglandin E by macrophages.54 More recent work shows by cytogenetic analysis that the macrophages in AML long-term culture adherent layers are part of the leukemic clone in some cases but that defective support of normal hematopoiesis is also observed, with layers lacking leukemic adherent cells.55 There is defective differentiation of normal CD34(+)CD38(–) cells but not the more committed CD34(+)CD38(+) cells,56 suggesting selective inhibition of primitive hematopoietic precursors.
Functional defects in stromal layers may also be due to abnormalities in cytokine production, in addition to the reduced M-CSF and increased TNF-α levels described previously. Constitutive expression of IL-1β, IL-6, and G-CSF transcripts was observed in AML but not in normal adherent layers.57 Leukemia inhibitory factor (LIF) protein levels were significantly raised in AML stromal layer supernatants compared with normal controls.58 The significance of raised LIF levels in this context is unclear, but in conjunction with marrow stromal cells, the factor is known to enhance the proliferation of CD34(+)CD90(+) cells.59
It is evident that the interactions of hematopoietic and stromal growth factors and inhibitors on normal and leukemic hematopoiesis in in vitro cultures are highly complex. Results from different studies may be difficult to compare because culture conditions and cellular composition of the stromal layers can vary significantly, especially for levels of stromal macrophages. The relative proportions of cellular components of primary stromal layers are rarely reported but may have a profound effect on cytokine profiles, making comparisons of studies with cloned stromal lines even more problematic. Caution should also be exercised in evaluating studies that report cytokine messenger RNA versus protein levels because of the uncertain significance of subliminal levels of the former.
Interaction of Leukemic Cells with Stromal Extracellular Matrix
AML cells can adhere to a wide variety of ECM components.23 As discussed above, AML blasts adhere to ECM fibronectin and laminin through the β1 (VLA-4, VLA-5, VLA-6) and β2 (LFA-1) integrins found on the leukemic cells.22,24 Antibody-blocking studies show that this only partially accounts for the adhesion24 and that other mechanisms, including CD44 binding, have been invoked.23 Many more CD44 variants are expressed on leukemic than on normal hematopoietic cells, and they may affect the interaction of AML cells with stroma.60 A 67-kd receptor present on CD14(+)CD11a(+) AML cells with monocytic morphology but absent on normal bone marrow cells mediates specific adhesion to laminin and represents a novel mechanism for the interaction of leukemic cells with the HM.61
The reduced apoptosis of AML cells documented in several in vitro models is not restricted to stromal cell contact-mediated mechanisms. The adhesion of c-kit(+) AML blasts to fibronectin is enhanced by SCF, which augments the fibronectin/VLA-5-mediated inhibition of apoptosis and increases leukemic cell proliferation.62
Evidence for Malignant Stromal Cells
Although stromal macrophages have been shown to be of leukemic origin,55 definitive studies demonstrating that nonhematopoietic stromal cells are part of the neoplastic clone are lacking. Moreover, marrow stromal cells from patients with the best-characterized multipotent stem cell disorder, Ph(+) chronic myeloid leukemia, are Ph negative.63 However, in a study of patients with myeloproliferative disorders heterozygous for glucose-6-phosphate dehydrogenase, the nonhematopoietic stromal cells from adherent layers of long-term marrow cultures were derived from the same clonal progenitors involved in the multipotent stem cell disorder.64 In contrast, the stromal cells were nonclonal in a patient with a restricted-type AML (clonal granulopoiesis and nonclonal erythropoiesis). Further investigation is needed with better clonal markers and methodologies that completely eliminate contaminating stromal macrophages to test this intriguing possibility, especially in light of recent work demonstrating stem cell plasticity. Nonetheless, the paucity of positive data makes the possibility that there are malignant stromal cells less likely; or at least these cells are at best probably uncommon.
Conclusion
It is evident that leukemic cells interact with the HM at many levels and mimic the action of normal early precursors to a variable extent, as summarized in Figure 1 and Table 4 . Like normal cells, AML blasts adhere to stromal cells and ECM components, but in contrast, they may receive additional protection from endogenous apoptotic mechanisms or apoptosis-mediated chemotherapy. In situ, leukemic cells may proliferate in response to any, or all, of the adhesive interactions with stromal cells, ECM components such as fibronectin and laminin, or local gradients of cytokines in the HM that are secreted by stromal cells, are generated by autocrine mechanisms, or are found in association with glycosaminoglycans (for example, heparan sulfate). Aberrant expression of CAMs on leukemic blasts may account for different patterns of trafficking and possibly the clinical presentation of AML subtypes. In these many steps there are opportunities for therapeutic intervention. One approach that merits further investigation is to develop ways to interfere with the suppression of the apoptosis of AML cells that is mediated in particular by ECM fibronectin.
II. Stem Cell Transplantation in Acute Myeloid Leukemia in the Younger Adult
Anthony H. Goldstone, MD,*
University College Hospital, Grafton Way, London WC1E 6AU, United Kingdom
Dr. Goldstone is a consultant to Roche and Novartis.
Despite the fact that 70-80% of patients with AML achieve complete remission (CR) most of them eventually relapse and die of the disease.1,2 Once remission has been achieved, further intensive therapy is needed to prevent relapse. Patients under the age of 60 have three main options after going into remission: intensive chemotherapy (IC), autologous stem cell transplantation (ASCT), or allogeneic stem cell transplantation (allo SCT) of some kind.3–,8 Patients with standard-risk disease have traditionally been referred for a matched sibling allograft if a donor is available and the patient’s performance status is adequate. In recent years, chemotherapy postremission has improved patients’ outcome, with a 50% 5-year overall survival (OS) and 40% disease-free survival (DFS) similar to that achieved with allo SCT, narrowing the differences between chemotherapy and such transplants8 (MRC AML 12, Burnett et al, unpublished data, 2002) (Figure 2). The high mortality of allograft (20%-25%), even today, is therefore making it less attractive in comparison with chemotherapy, and selection of patients to be given allografts in first remission (CR1) should be done very carefully.
Risk Group Designation
Although all patients included in these studies were diagnosed with AML, there is a considerable variation in their risk of relapse. Cytogenetics is now considered the most powerful single prognostic factor.9,10 Whilst patients with t(15;17) or chromosomal abnormalities involving the core binding factor [t(8;21) and inversion 16] are classified as having a favorable prognosis, with approximately 30% risk for relapse, patients with abnormalities in chromosome 5/7/3(q) or multiple chromosomal abnormalities have an approximately 75% chance of relapse (Table 5 ).9,11 However, most patients do not belong to these two categories and are classified as having standard-risk disease (5-year OS = 43%).9,11
Recent studies have indicated that an internal tandem duplication (ITD) in the FLT3 gene may adversely affect clinical outcome in AML patients.12
Prevention of Relapse in Young Patients
A significant reduction in relapse rate (RR) has been observed since the introduction of intensified postremission therapy.1,3 The Cancer and Leukemia Group B (CALGB) randomly assigned 596 patients in CR1 to receive 4 courses of cytarabine at 1 of 3 doses.1 High-dose cytarabine (18 g/m2/course) was demonstrated to be superior to 2 g/m2/course, with a DFS of 44% versus 29% (P = 0.003) and an OS of 52% versus 40% (P = 0.02).1
A different consolidation regimen, containing no more than 1 g/m2 cytarabine rather than high dose, has been successfully used by the Medical Research Council (MRC) AML 10 trial (DFS and OS were 43% and 40%, respectively).2,3
The number of postremission chemotherapy courses required is unresolved. Recent data of the MRC AML 12 trial seem to show no advantage for 4 consolidations compared with 3.13
Main prospective trials
EORTC/GIMEMA AML 8 trial (1986-1991)
The European Organization for Research and Treatment of Cancer (EORTC) and the Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto (GIMEMA) Leukemia Cooperative Groups5 carried out the first published prospective randomized study designed to review the best consolidation therapy for AML patients (Table 6 ).5
AML patients 10 to 45 years old who entered CR after 1 or 2 courses of induction were then treated with intermediate cytarabine dose (6 g/m2/course + amsacrine, 1 course), followed by allograft if matched related donor was available, or randomized to ASCT versus chemotherapy (cytarabine 16 g/m2/course + daunorubicin) if no donor was available. There was no significant survival advantage for allo SCT compared with chemotherapy or ASCT (4-year OS for allo SCT = 59%, ASCT = 56%, IC = 46%). However, both allo SCT and ASCT seemed to have a better antileukemic effect compared with chemotherapy, reflected by reduced RR and improved DFS (Tables 7 and 8). It seems that these results reflect the relatively poor outcome observed with chemotherapy, rather than being a true “superior effect” of the transplant (alloSCT/ASCT) itself.
EORTC/GIMEMA AML 10 trial (1993-1999)
Building on their previous study, the EORTC/GIMEMA conducted another study, aiming to clarify the role of ASCT versus allo SCT. Patients who achieved CR received IC followed by allo SCT if they had a matched related donor, or ASCT if no matched related donor was available.7
Nearly half of the patients without a donor had ASCT. A donor versus no donor analysis revealed a reduced RR with increased DFS in the donor group (RR: 31.2% vs 52.9%, P = 0.0001; DFS = 51.4% vs 41.2%, P = 0.046). A survival advantage was most noticeable in poor-risk patients who had a donor (4-year DFS = 43.8% vs 19%; OS = 50.4% vs 27.7%).
GOELAM trial
In the Groupe Ouest Est Leucemies Aigues Myeloblastiques (GOELAM) trial, patients with de novo AML who entered CR were assigned to allograft if they had a matched related donor and were less than 40 years old.6 All other patients were consolidated with 1 chemotherapy course of high-dose cytarabine and anthracycline. At this stage, patients were randomized to receive ASCT, or a second course of IC, containing amsacrine and etoposide (Table 6).
Despite the relatively low mortality rate (22%) reported in those assigned to allograft, there was no increase in OS compared with patients without a donor, partially because of the high RR (37%) observed in the donor group. There were no significant differences in DFS and OS between patients assigned to ASCT versus IC (Table 8). However, ASCT was associated with high myelotoxicity, especially prolonged thrombocytopenia (109.5 days with ASCT vs 18.5 days with IC).
MRC AML 10
Between 1988 and 1996, 1966 patients with de novo or secondary leukemia were recruited to the MRC AML 10 trial. All patients were treated with 4 chemotherapy courses and were then assigned to allo SCT if they had a matched related donor (n = 419) or randomized between ASCT (n = 190) and no further treatment (n = 191) if no donor was available (Table 6).3,8 Analysis of donor versus no donor groups revealed a significantly higher transplant-related mortality (TRM) rate in the donor group: 19% versus 9% (P < 0.001). Twenty-four percent (62/255) of the patients who received allo SCT died in remission, compared with 17 of 156 (11%) who had a donor available but did not receive an allograft.8 Relapse risk was significantly lower and DFS was significantly higher in the donor group compared with the “no available donor” group (RR: 36% versus 52%, P = 0.0001; DFS: 50% versus 42%, P = 0.001).8 Nevertheless, there was no survival advantage for allo SCT compared with other treatments (7-year OS = 56% for donor group vs 50% in no donor group, P = 0.1).8 However, it seems that patients with standard-risk cytogenetics who are younger than 35 do have a survival advantage with allo SCT.8 This favorable outcome observed with allo SCT might result from patient selection bias (selection of patients with a favorable biology of disease who succeed in remaining in CR during the chemotherapy courses preceding the transplant) rather than being due to the superiority of transplant.
Intent-to-treat analysis assessing the outcome of ASCT versus no further treatment showed a significant reduction in RR in patients assigned to autograft (37% vs 58%, P = 0.0007) (Table 8).3 Despite the increased DFS in patients assigned to ASCT (DFS = 54% vs 40% with no further treatment, P = 0.04, Figure 3 ), there was no increase in OS, reflecting the higher mortality rate (12%) observed with autologous transplantation (P = .008).3 The main cause of death was impaired hematopoietic reconstitution.
MRC AML 12 trial
In MRC AML 12, researchers attempted to clarify whether 5 courses of therapy were better than 4 and whether the last course of therapy should be transplant.13
Patients with de novo or secondary AML were randomized to receive MAE (mitoxantrone, cytarabine, etoposide) versus ADE (cytarabine, daunorubicin, etoposide) [replaced from 1998 by S-DAT (daunorubicin, cytarabine 100 mg/m2, thioguanine) versus H-DAT (daunorubicin, cytarabine 200 mg/m2, thioguanine) with or without all-trans retinoic acid (ATRA)] as first chemotherapy course (Table 2). Patients who remained in CR after the third course of chemotherapy underwent a second randomization into 4 versus 5 courses of treatment and transplant versus chemotherapy.13 Those who were randomized to transplant and had a matched related donor were assigned to allograft, whereas patients without a donor were assigned to peripheral blood stem cell transplantation (PBSCT) or bone marrow stem cell transplantation (BMSCT).
First results of AML 12 (still unpublished) show reduced relapse risk in patients having a donor. However, no significant survival advantage for having a donor has been observed in any of the 3 risk groups (Figure 2). There was no increase in OS in patients assigned to autograft, and 5 chemotherapy courses in total appeared to provide the same results as 4.13
The Intergroup (SWOG, CALGB, ECOG) trial
In the Intergroup (Southwestern Oncology Group [SWOG], CALGB, Eastern Cooperative Oncology Group [ECOG]) trial, high-dose consolidation therapy was compared with ASCT and allo SCT in CR1.4 Patients who achieved CR were treated with modest-dose cytarabine (500 mg/m2) followed by allo SCT if a matched related donor was available. Patients without a donor were randomized to receive a purged ASCT versus one high-dose cytarabine course (Table 6).
Allo SCT appeared to provide the best antileukemic effect, associated with a RR of 29%, compared with 48% in patients assigned to ASCT, and 61% for patients assigned to chemotherapy. However, the lower RR observed with allograft was reversed by a high mortality rate, approaching 25% (TRM = 25% with allo SCT vs 14% for ASCT and 3% for IC). Despite the high RR observed with chemotherapy, chemotherapy appeared to be superior to ASCT or allo SCT (Table 7). It seems that the low delivery rates of assigned treatments, a longer duration from CR to transplantation compared with IC and the relatively high TRM observed with high dose therapy (HDT) (especially higher than expected for patients assigned to autograft) might explain the unsatisfactory results observed with HDT rather than reflecting a real advantage of IC.
Main Problems Interpreting Results of Prospective Trials
There remain some problems with interpreting the results of major prospective AML trials.14 Patients who receive allo SCT are clearly selected, because a proportion of patients with a matched sibling donor do not receive a transplant. Patients may be excluded from transplant because of early relapse/death, previous complications with chemotherapy, or other medical problems. It is not possible to predict the direction of such biases14 and indeed they are at the core of problems with interpreting registry data.
On intent to treat basis, “crossover” between arms and/or a failure to receive intended treatment, may in some circumstances radically underestimate differences between arms, if happening to a significant degree.
Both compliance and randomization were quite poor in most prospective trials (Table 6). As a result, both beneficial and harmful effects might be underestimated.
Allo SCT is usually delayed, which might lead to greater selection of patients with favorable disease who remain in CR until transplant.
A large number of patients are needed for a difference in the efficacy between postremission therapy options to be detected. For example, to detect a 10% difference in survival from 40% to 50% (P = 0.05 with 90 power), 1000 patients are needed.14
Allo SCT in AML
With chemotherapy producing a less than 20% chance of survival before the early 1980s, durable survivals up to 50%, with a low relapse risk of 15-25%, were reported in patients receiving allografts in CR1.15,16 Today, the DFS and OS in nontransplanted patients begin to achieve this rate of durable survival and make such treatment comparable to that achieved with allograft3,8 (MRC AML 12, unpublished data).
Allo SCT in the main prospective trials
All trials confirmed allo SCT to be the best antileukemic treatment, associated with a relapse risk of 24%-36% compared with 46%-61% observed with ASCT/IC.3-8,17 However, almost all prospective studies failed to show an improved OS in patients assigned to allo SCT (see Table 7).
The MRC AML 10 trial observed a survival advantage for patients treated with allograft compared with patients treated with IC who had no available donor. 8 However, the survival of patients who had a donor but were eventually treated with only IC was inferior to the survival of not only allografted patients but also the “no donor” group. These data indicate that patients who eventually received the transplant were biologically selected to have a favorable prognosis, as patients with poorer prognosis did not get the transplant. However, the EORTC/GIMEMA AML 10 trial has recently reported a higher survival rate in poor-risk patients assigned to allograft (OS = 50.4% versus 27.7%).7
Summarizing the data regarding allograft in CR1 in AML patients remains difficult:
None of the trials is truly prospective with full biological assignment based on donor availability as surrogate for intent-to-treat analysis.
Pretransplant chemotherapy varies in its number of courses in some trials (Table 6). This variability in the number of courses might affect transplant outcome in relation to both toxicity and time to treatment bias.
All studies had problems in delivering the assigned treatment (Table 6). This might underestimate both its efficacy and toxicity.
The superiority of allo SCT depends upon comparison with the best available IC, but the best available IC was not always used in every trial (Table 6).
Upper age limit for transplant will affect outcome, as toxicity increases with age, probably more than with chemotherapy treatment alone (Table 6).
Most of the major studies were initiated more than 10 years ago. The currently improved HLA-matching stem cell transplant technology and supportive care may now make many of the toxicity figures meaningless. PBSCT may also reduce relapse risk.18,19 The problem remains, however, that big studies with a large number of patients take a number of years to conduct, and during that period various aspects of treatment can change radically.
Varying RRs of different risk groups mean that therapy should be tailored according to each individual patient’s risk.
BMSC or PBSC for Allo SCT?
The main reason for the increasing use of PBSC relies on the rapid hematopoietic recovery observed with PBSC compared with BMSC.18,19 A few studies have also reported improved immune reconstitution using PBSC.18,20 However, PBSCT might be associated with increased incidence/severity of acute graft-versus-host disease (AGVHD) and chronic graft-versus-host disease (CGVHD).21 Champlin et al, on behalf of the European Group for Blood and Marrow Transplantation (EBMT) and the International Bone Marrow Transplant Registry (IBMTR) working committee, have retrospectively compared the outcome of patients treated with PBSCT (n = 288) and BMSCT (n = 536).21 Incidence of CGVHD, but not AGVHD, appeared to be higher in patients who received PBSCT. Patients with advanced disease at transplantation (AML patients in second remission [CR2] and chronic myelogenous leukemia patients in accelerated phase) seemed to have reduced TRM and increased leukemia-free survival (LFS) when receiving PBSC compared with BMSC.21 However, these advantages were not observed in AML patients receiving PBSCT in CR1.21 Conversely, Russell et al have reported an improved DFS in patients who received PBSCT in CR1.22
Randomized trials confirmed an improved engraftment with PBSCT compared with BMSCT.19 The Seattle Group randomized 172 patients with hematological malignancies (including 21% AML patients) to receive PBSCT versus BMSCT.19 Despite the improved DFS observed with PBSCT compared to that with BMSCT (2-year RR: 14% vs 25%, P = 0.04, DFS: 65% versus 45%, P = 0.03), there was no significant increase in OS (OS = 66% versus 54%, P = 0.06). TRM, though lower in patients treated with PBSC, was still high (21% versus 30%, hazard ratio 0.7; 95% confidence interval 0.38-1.28; P = 0.24). In contrast to the higher incidence of CGVHD reported in some retrospective studies,21 there was no significant difference in the incidence of AGVHD/CGVHD in this prospective trial. Further studies comparing PBSCT and BMSCT and allowing longer follow-up are needed before any one stem cell source can be deemed superior.
T-cell depletion
GVHD is a major cause of mortality and morbidity after allo SCT, and removing the T lymphocytes from the donor bone marrow can decrease the incidence of both. However, unselected T-cell depletion may theoretically increase RR (preventing the graft-versus-leukemia effect) and enhance treatment-related infections due to delayed immunological recovery.
Low-intensity stem cell transplantion
Low-intensity stem cell transplantation (LI SCT) is being increasingly used, aiming to exploit the curative potential of allo SCT by inducing graft-versus-tumor effect without the morbidity and mortality associated with conventional transplantation. Low-intensity SCT is less toxic and therefore may be considered for some patients who are otherwise not eligible for conventional allogeneic transplant. However, its efficacy in AML patients has still not proven to be as good as that of high-intensity allo BMT. The EBMT reported on 69 patients (median age 51) treated with LI BMT for AML or myelodysplastic syndrome (MDS).25 More than half of the patients had refractory/relapsed disease at transplantation. Graft failure was more frequent than observed with conventional allo SCT, though 77% achieved > 95% donor chimerism. Patients’ outcome was highly dependent on disease status at transplantation: 1-year TRM, RR, and OS were 47%, 30%, and 41% for the whole group, compared with 17%, 21%, and 67% in patients transplanted in CR (CR1 or later). However, follow-up was still short at time of publication.25
Peggs et al have recently reported 44% progression-free survival (PFS) and 53% OS (median follow-up, 18 months) in 24 patients aged 18-60 years (median 47) treated with low-intensity matched related/unrelated SCT (total body irradiation, melphalan, fludarabine, Campath-1H) for MDS/AML (n = 17).26 DFS and OS were higher in AML patients transplanted in CR1 (n = 15), approaching 57% and 62%, respectively.26 Longer follow-up and prospective comparison with IC are needed in order to define the role of LI SCT in selected AML patients. The MRC AML 15 trial intends to allow the possibility of LI SCT in patients aged 35-45 years who have a matched related donor, while suggesting conventional transplant for the younger recipients.
Autologous Stem Cell Transplantation
ASCT in CR1
On the assumption that HDT has value in AML, several groups conducted nonrandomized trials using ASCT as consolidation therapy in CR1.27–,29 Encouraging results, observing a reduced RR with improved DFS compared with chemotherapy, were reported.27– 29 Patient selection, however, might have played a role.
Does stem cell purging improve patient’s outcome?
Despite the superior DFS observed with an ASCT compared with chemotherapy,27–,29 RR remained higher than observed with allo SCT and the main cause of death was recurrence of disease. Some investigators have tried to purge stem cells pretransplantation, aiming to reduce RR and improve OS.28,30,31However, results were variable.4,30,31 Furthermore, pretransplant purging may carry a risk of loss of accessory and progenitor cells, resulting in delayed hematopoietic and immunological reconstitution.
ASCT in prospective randomized trials
One of the main unsolved questions is whether there is any place for ASCT in CR1 and which patients (if any) should be transplanted. All three prospective trials designed to compare IC with ASCT 4–,7 observed a reduced RR in patients assigned for autograft, reflecting its superior antileukemic effect compared with chemotherapy (Table 8).4–,7 The MRC AML 10 trial, comparing ASCT with no further treatment, reported a significantly increased DFS in patients assigned to ASCT (Figure 3).3 Failure to deliver assigned treatment, observed with all trials (Table 6), might underestimate the real efficacy of ASCT. All prospective trials failed to show a survival advantage in patients assigned to autograft compared with chemotherapy. The high mortality rate reported with ASCT (12%) offsets the antileukemic advantage provided with autograft. In addition, some patients who relapsed postchemotherapy were salvaged with HDT.3,5 However, the TRM associated with ASCT is no longer today significantly higher than that observed with IC. A reduced TRM might therefore be translated into increased OS.
ASCT for patients in second or later CR
ASCT might have a place in rescuing patients who have relapsed postchemotherapy, with up to 20-50% relapse-free survival in selected patients.3,5,32– 34
Linker et al34 have recently reported a 5-year DFS of 54% in patients with advanced leukemia (patients with primary induction failure who remitted with salvage therapy/patients in CR2 or later CR) who were treated with high dose cytarabine/etoposide consolidation, followed by an ASCT.34
It is still unclear if patients in CR2 or later without a donor should be referred to matched unrelated donor (MUD) transplantation instead of ASCT. A prospective comparison of MUD versus ASCT in this situation is very difficult because time to treatment might be delayed and some MUD patients may not be in true remission while others, proposed autograft, might fail to obtain an adequate stem cell harvest.
BMSCT or PBSCT in ASCT?
Retrospective studies observed a faster hematopoietic recovery, associated with a lower morbidity and mortality (TRM = 5%) in patients treated with PBSCT compared with BMSCT.28,29
Most studies did not show any differences in RR or DFS between stem cell sources.35–,37 The EORTC/GIMEMA AML 10 trial randomized patients with no available donor to BMT (n = 146) or PBSCT (n = 146).37 There were no significant differences in DFS and OS between the two groups (4-year DFS = 49.8% with BMSCT vs 42.6% with PBSCT, P = 0.33; OS = 55% vs 55.3%, respectively).37 It is still unclear whether PBSCT provides any other advantage compared with BMT, except of facilitating stem cell engraftment.
Outcome of Allo SCT in Different Risk Groups
Good-risk patients
All prospective studies3,5–,8 except the Intergroup trial4,10 failed to show a survival advantage with allo SCT in CR1 in patients with favorable cytogenetics. It appears that the Intergroup result might reflect a random result rather than a genuine tendency, because of the small number of patients included.
Acute promyelocytic leukemia (APL) patients, but not patients with t(8:21) or inversion 16, were reported to have a significantly lower RR with allograft in CR1 (donor vs no donor analysis: 22% vs 43%, P = 0.02).8 However, it seems that since the introduction of ATRA, nontransplant therapy can provide the same good result. The only study that observed a survival advantage in good-risk patients treated with autograft was the Intergroup study.4,10 However, this “superior” outcome might actually reflect the poor results achieved with chemotherapy.4 It now seems that there is no place for allo SCT or ASCT in CR1 in patients with favorable-risk cytogenetics.
Standard-risk patients
Both the MRC AML 10 trial3,8 and the EORTC/GIMEMA AML 10 trial7 reported reduced RR and improved DFS in patients with standard-risk disease assigned to allo SCT. However, it is still unclear whether allo SCT can improve patients’ OS (MRC AML 12, unpublished data).7
There is still significant heterogeneity in this group of patients and RR is influenced (independent of cytogenetics) by response to first induction and the presence of the FLT3 mutation. It is possible that some of these standard-risk patients (e.g., those who express the FLT3 mutation or failed to remit with first course of induction) will do better with allo SCT, while others will gain no advantage from being transplanted.
Poor-risk patients
Researchers from the EORTC/GIMEMA AML 10 trial have recently reported their current results.7 When patients are divided into risk subgroups based on their cytogenetics, it seems that patients with unfavorable cytogenetics get the maximal benefit from having allo SCT compared with ASCT or IC.7 Of interest, the EORTC/GIMEMA AML 10 study considered all patients without a favorable/normal karyotype as having poor-risk disease (Table 5). However, the MRC AML 10,8 which failed to show an advantage for allograft in poor-risk patients but showed one for the standard-risk group, included approximately half of these patients with unfavorable cytogenetics in the standard-risk group rather than in the unfavorable-risk one.
Other Options for Transplantation for Patients Without a Matched Related Donor
Matched unrelated donor transplantation (MUD)
MUD in CR1. Despite the controversy about the advantage of matched related donor allograft in CR1 in poor-risk patients,7,8,10 the very grim prognosis observed with chemotherapy (less than 30% DFS) might justify using MUD in CR1 in this group of patients. However, there is currently little evidence that any kind of allo SCT can cure large numbers of these poor-risk patients.
MUD in CR2. Patients with unfavorable cytogenetics who achieve a second CR and have no matched related donor are often referred to MUD SCT. However, MUD in CR2 in patients with favorable-/standard-risk cytogenetics remains controversial and its superiority compared with ASCT is still questionable.38–,40 Lazarus et al, on behalf of the IBMTR, have retrospectively compared the outcome of AML patients (CR1/CR2) treated with MUD versus ASCT between 1989-1996.39 Three-year LFS was 33% in MUD patients versus 40% with an autograft. However, long-term side effects were significantly higher in MUD patients and selection bias is unknown.
On the basis of these incomplete retrospective data, it may be reasonable to consider MUD in young patients with adverse cytogenetics and short CR1s who have a matched unrelated donor (at least 10 antigen matching).40 Conversely, patients older than 40 with a long CR1 may do better with ASCT, if their disease is in genuine remission and enough cells can be harvested.40
T-replete versus T-depleted MUD. The reduced GVHD-related mortality achieved with T-cell depletion is often balanced by increased RR and overwhelming infections.
In contrast to patients with some other malignancies, AML patients may achieve a survival advantage with T-depleted marrows compared with T-replete ones,40 justifying T cell depleted MUD in selected AML patients. Nevertheless, data justifying this are still scanty and further studies are needed to support this strategy.
Haploidentical BMT
Haploidentical BMT is an option for patients who do not have a matched related donor (approximately 70% of patients). The historical data concerning haploidentical BMT in AML patients were disappointing. These disappointing results might be attributed to patient selection for transplant (advanced disease) as well as to a high rate of transplant-related complications, mainly GVHD, which in turn was replaced by a high incidence of graft failure as T-cell depletion has been introduced to prevent GVHD. T-cell depletion by itself was associated with delayed immune recovery, resulting in high incidence of severe infections. However, it seems that recent modifications have succeeded in making some progress. Stem cell megadose (106 CD34 cells/kg) is essential to overcome the HLA barrier in full haplotype-mismatched transplants.41 Further reduction of T-cell dose infused reduces the frequency and the severity of GVHD significantly. Posttransplant granulocyte colony-stimulating factor (G-CSF) appears to interfere with natural killer (NK) cell recovery and has therefore been excluded.42 Donor’s NK cell alloreactivity, a unique phenomenon of mismatched transplants, appears to play an essential role in preventing relapse and supporting stem cell engraftment.43 A recent update from Perugia suggests that the current morbidity and mortality in AML patients having haploidentical BMT is not higher than reported with matched allogeneic BMT.44 Event-free survival in high-risk patients transplanted in CR1/CR2 approached 45%, with an RR of less than 15%.44 A donor versus recipient NK cell alloreactivity is essential for achieving graft-versus-tumor effect and may therefore become a major criterion for donor selection in mismatched SCT.
Acute Promyelocytic Leukemia
None of the large randomized trials showed a survival advantage with ASCT/allo SCT in CR1 compared with the outcome achieved with ATRA-containing regimens. However, for patients in second CR, there might be an advantage with both kinds of transplants.45 An achievement of molecular remission pre-autograft is essential and is associated with a high cure rate.45,46 Conversely, patients transplanted with evidence of minimal residual disease (MRD) (morphological remission without molecular remission) have high risk for relapse46,47 and should therefore be considered for an alternative therapy such as allo SCT or arsenic trioxide.45,47 Postautograft maintenance therapy with ATRA might further reduce RR, although this has to be confirmed.
Options for Treatment in Elderly Patients
The age-related differences in biology of disease partly explain the poor outcome observed in patients older than 60 than in younger patients. However, undertreatment might also contribute to these inferior results.
Postremission therapy with high-dose cytarabine failed to improve the outcome of patients older than 60 and has been associated with high toxicity.1
The advantage of ASCT in CR1 is not proven either, though some investigators have reported an improved survival with ASCT;48,49 however, a selection bias might be involved. Allo SCT, being associated with higher TRM in elderly patients (especially GVHD related), is even less feasible. Deeg et al50 have recently reported a nonrelapse mortality of 39% in 50 MDS patients (16 with transformation to leukemia) aged 55-66 years (median 58.8) treated with conventional allo SCT. However, encouraging results with low-intensity SCT in elderly AML patients have recently been reported.26
It appears that stem cell transplantation (ASCT or allo SCT), being often associated with high toxicity, will not be the routine treatment in elderly patients. Overcoming the drug resistance that frequently exists in elderly patients with AML has remained one of the main challenges to treating them.
Minimal Residual Disease
Detection of MRD posttherapy, aiming to identify patients who are at higher risk for relapse, remains a major challenge.51,52 Molecular methods (polymerase chain reaction [PCR]),51,52 as well as immunological methods (multi-parametric flow cytometry analysis)52,53 have been employed. Whereas immunophenotype analysis can be informative in 80%-85% of AML patients, molecular analysis can be useful in less than 30% of patients, as only the minority of AML patients express a traceble molecular marker (e.g., AML1/ETO, PML/RARα, and possibly the FLT-3 mutation).51,52 There are some patients whose leukemic cells present chromosomal abnormalities that can be monitored with fluorescence in situ hybridization (FISH).51 However, FISH is usually less sensitive than molecular monitoring, and the sensitivity of it may be inadequate.51,52 Furthermore, 30%-50% of AML patients have normal karyotype.
The significance of a positive MRD finding is not always clear and a different level of residual disease seems to be significant for different types of AML.54,55 Tobal et al reported a tenfold difference in MRD levels between patients in long-term remission from APL54 compared with patients with t(8;21).55 Molecular remission in APL patients seems to be associated with long-term DFS, whereas failure to achieve a molecular remission does not.45–,47,56,57 In contrast, a positive molecular result in patients with t(8;21) who obtained clinical remission is not necessarily associated with impending relapse.51,58 APL patients who achieved a molecular remission have an excellent prognosis having IC only.45– 47 However, data are still too scanty to apply this strategy to other AML subtypes, and further studies including quantitative monitoring should be investigated.
Conclusions and Future Directions
Despite the contributions of large prospective trials comparing the efficacy of allo SCT with that of IC or ASCT, it is still unclear what the best postremission therapy is for AML patients.
ASCT in CR1
It appears that patients in the favorable-risk group do not require HDT in CR1, as their outcome with chemotherapy is excellent.8 There is no clear evidence that standard-risk patients are doing better with allo SCT than with IC either (MRC AML 12 unpublished data, Figure 2). With the lack of major benefit for allo SCT, the poorer quality of life often observed post allograft should be considered.59 Patients will need to decide whether increasing their chance of survival by a few percentage points is worth the potential long-term morbidity. Also, issues of infertility will be perceived differently by the patients in different age groups and those with different family circumstances.60
The role of allo SCT in CR1 in poor-risk patients remains unclear,8 although some studies have showed some advantage compared with chemotherapy.4,7,10
However, all these large prospective studies have used BMSC, whereas recent studies have shown a possible reduced RR with PBSCT compared with BMSCT.18,19 Prospective studies comparing PBSCT with BMSCT are needed to clarify these issues.
Better understanding of prognostic factors (e.g., FLT3 status, MRD status) might help in selection of the patients who will benefit from allo SCT. The number of chemotherapy courses needed pretransplantation is unclear61 and might be influenced by patients’ risk group (including evaluation for MRD) and planned HDT.
New kinds of allo SCT based on reduced chemotherapy doses (LI SCT) or T-cell depletion allograft may succeed in reducing TRM observed with conventional allo SCT and allow transplantation to be considered in patients who are not eligible for conventional allograft, including elderly patients.
ASCT/IC in CR1
The role of ASCT in CR1 is not clear. All prospective trials showed reduced RR in autografted patients. However, survival was not higher than observed with chemotherapy, mainly because of the high TRM reported with ASCT. A current prospective study, including a large number of patients, comparing IC with PBSCT and BMSCT is needed. Improving the outcome achieved with chemotherapy remains one of the main challenges to be addressed by current studies. Attempts to overcome chemotherapy resistance, which is most noticeable in elderly patients, need to be ongoing.
Future studies
In the absence of any clear evidence for the role of both allo SCT and ASCT in CR1, the main trial groups have designed different directions for future research. ECOG is soon starting a new treatment protocol retaining the option for autotransplant compared with allo SCT and chemotherapy. Chemotherapy intensification will be further investigated, including an increased dose of daunorubicin and combining anti-CD33 with chemotherapy (Figure 4 ).
In contrast, the MRC AML15 trial has decided to omit ASCT, having seen no evidence yet of a survival advantage with ASCT. Patients with standard- or poor-risk disease who are younger than 35 and have a matched related donor will receive a PBSC/BMSC allograft. Their outcome will be compared with that of patients with no available donor, treated with further chemotherapy courses (Figure 5 ). Patients 35-45 years old who have a matched related donor can receive a conventional allograft, or an LI BMT, depending on investigator or patient choice (but not randomized).
Hopefully, new ongoing prospective trials will give a better idea about the current value of allo SCT compared with ASCT and IC in adult AML patients. However, if nontransplant options continue to improve at a greater rate than transplant options, then selection of appropriate patients for transplant will become more and more difficult.
III. Biologic and Genetic Risk Assessment of AML in the Genomic Era
Cheryl L. Willman, MD*
UNM Cancer Research and Treatment Center, 2325 Camino de Salud, N.E., Albuquerque, NM 87131-5636
Acknowledgment of Research Support: DHHS NIH U01CA32012, U01CA60433, U01CA30969, R01CA186026 and the W.M. Keck Foundation, Los Angeles, Calif.
Acknowledgments: I would like to thank my colleagues associated with the Southwest Oncology Group and the Pediatric Oncology Group/Children’s Oncology Group, who with many colleagues throughout the world have made significant contributions to both scientific and clinical advances in leukemia. I would also like to thank my colleagues and research team at the University of New Mexico (UNM) Cancer Research and Treatment Center, the UNM High Performance Computing Education and Research Center, and Sandia National Laboratories, whose collaborations have been essential and critical for our genomic studies of acute leukemia.
In the past two decades, important scientific advances have yielded new insights into the epidemiologic, genetic, and biologic features of the AMLs.1–,6 Yet despite these scientific advances, the majority of patients affected by AML still die of their disease.6 With the exception of acute promyelocytic leukemia (APL), we have not yet succeeded in translating our scientific discoveries into more effective treatments for the majority of AML patients. While therapeutic intensification and improved supportive care have led to gradual improvements in outcome in children and younger adults with AML (particularly those with more favorable cytogenetic abnormalities), overall survival in this age group still approaches only 50%.6 In older individuals (> 55-60 years) and in secondary AML patients, in whom resistance to current therapies and an increase in unfavorable cytogenetics characterizes the disease, the outlook is even more dismal, with overall survivals of 10% to 15%.7–,14 As the majority of individuals affected by AML are in this older age group,15 we thus lack effective therapeutic approaches for the majority of patients with this disease. New laboratory and clinical approaches that can be successfully translated into more effective diagnostic and therapeutic strategies are desperately needed for AML. We must define the most important questions to advance our knowledge and focus on the discovery of new therapeutic strategies for AML patients with high-risk as well as low-risk disease. Molecular genetics and gene expression profiling hold promise to improve AML disease classification systems, to model gene expression profiles associated with chemoresistance or response to various agents, and to identify novel therapeutic targets.16– 23 This section will briefly review preliminary accomplishments and ongoing studies in this field of investigation.
AML Prognostic Factors 2002: Age, Antecedent Disease, Cytogenetics
From the past two decades to the present day, the most important prognostic factors in AML have remained: (1) patient age; (2) presenting white blood cell count; (3) whether the AML presents clinically as de novo disease or secondary to antecedent myelodysplasia (MDS) or leukemogenic therapies (therapy-related AML, t-AML); and (4) the presence of specific cytogenetic abnormalities, usually clustered as “favorable,” “intermediate,” or “unfavorable” (Table 9 ).1,3,6 While AML accounts for only 13% to 14% of leukemia cases in the first 10 years of age (the majority being acute lymphoblastic leukemia or ALL), AML constitutes nearly 36% of the leukemia cases in older children.15 In adults, AML rates begin to rise exponentially after 50 years of age; age-specific incidence rates are 3.5 per 100,000 in adults 50 years of age, increasing significantly to 15 at age 70 and 35 at age 90. The mean age of AML in the United States is 63 years. Currently accounting for 10% to 20% of all AML cases, the incidence of secondary AML appears to be increasing due to the use of DNA damaging agents in cancer therapy at higher dose intensities and an increased length of survival for many cancer patients.24
Each of the broadly defined groups of AML (AML in infants and children, AML in younger adults, AML in older individuals, and secondary AML) is characterized by distinct but overlapping cytogenetic and molecular genetic abnormalities (Table 9). Although “favorable” cytogenetic abnormalities [such as t(8;21), inv(16), or t(15;17)] are more frequently seen in children and younger adults less than 55 years of age, it should be noted that these “good prognosis” cases still constitute a minority (< 35%) of the AML cases seen in this age group. In contrast to AML in younger patients, AML in older individuals is more frequently associated with unfavorable cytogenetic abnormalities (such as 7, 7q, 5, 5q, or complex karyotypes) and other poor prognosis biologic features (trilineage dysplasia suggestive of antecedent myelodysplasia or marrow injury and an increased frequency of intrinsic drug resistance).7–,14,25 The significantly poorer outcome seen in older AML patients is, in part, explained by a higher frequency of these unfavorable cytogenetic abnormalities and biologic features.7– 14 While younger and older AML patients define the extremes of response, the majority of patients affected by AML in all age groups fall into the “intermediate” cytogenetics category, in which risk classification and stratification are currently ill defined. While large clinical trials have demonstrated that AML-associated cytogenetic abnormalities confer important prognostic information, striking differences in therapeutic response and outcome may still be observed in leukemias with the same cytogenetic profile, implying that other more subtle genetic abnormalities and functional activation or inactivation of critical cellular pathways (signal transduction abnormalities, apoptosis, drug resistance, angiogenesis, cell cycle regulation, DNA repair) also impact disease biology and therapeutic response. Although all of these critical cellular pathways provide potentially important therapeutic targets for AML, we must determine how to best target genetically and biologically heterogeneous AML patients to each specific therapy. While karyotype provides critical prognostic information for AML patients in the context of current therapeutic approaches, it remains to be determined whether morphologic and cytogenetic classification schemes are the best approach to the classification of AML, particularly when considering targeted therapy. This is a question that can be directly tested using gene expression profiling.
Gene Expression Profiling in AML
Many investigators throughout the world are engaged in the use of microarray technology to derive gene expression profiles in the acute leukemias and other diseases. The goals of many of these studies are to understand the intrinsic biology of these heterogeneous diseases, to determine whether expression profiling can improve diagnosis and risk classification of the leukemias, to determine if expression profiles can be derived that predict for clinical outcome and response or resistance to current therapies, and to use expression profiles to identify new therapeutic targets. Such goals will require obtaining gene expression profiles on large, statistically designed cohorts of well-characterized leukemic patients in whom detailed cytogenetics, biologic covariables, and clinical outcome parameters are known and available. As described in several reviews and original reports,16–,23 gene expression profiles may be derived using both oligonucleotide or cDNA microarray technology. Methods have been derived that allow for the amplification and hybridization of small amounts of RNA,26 allowing gene expression profiles in AML and normal hematopoietic stem and progenitor cells to be compared. Although gene expression profiling studies in leukemia and MDS are still in their infancy, several preliminary studies have been reported. Miyazato and colleagues19 isolated AC133 surface marker-positive hematopoietic stem cells from 5 AML, 2 secondary AML, and 3 MDS patients; developed cDNA libraries; and compared gene expression profiles on custom oligonucleotide arrays containing approximately 1100 gene sequences. Although these authors attempted to identify genes more strongly associated with MDS (Dlk, Tec, inositol 1,4,5-triphosphate receptor 1) and AML (solute carrier family members, opioid receptor delta 1, and the leptin receptor), the expression of each of these genes was very heterogeneous and did not appear to precisely distinguish these related diseases. Focusing on AML, Guzman and colleagues20 compared the expression of 1400 genes in CD34+/CD38 cells isolated from 7 AML patients and from 3 normal bone marrow controls. Two tumor suppressor genes, IRF1 and death-associated protein kinase (DAPK), and several other genes (AML1, Af-4, EWS, Ikaros, and STAT6) were shown to be overexpressed in all AML progenitor samples relative to controls. Whether the expression of these genes can be used as markers of AML vs normal progenitor cells remains to be determined in larger confirmatory studies.
Not surprisingly, the results and conclusions from each microarray study for gene expression profiling depend on the characteristics of the patient sample set, how the questions to be addressed using microarrays were posed, the available clinical and laboratory data, and the methods employed for computational analysis of gene expression data.23 To date, the computational analysis of gene expression has centered on two different approaches. Unsupervised learning approaches, often termed “class discovery” or “clustering,” are used to uncover inherent “clusters” or biologic cohorts within a data set. Such “agglomerative” algorithms may be used to cluster a group of patients based on the similarity of their aggregate expression profiles or to alternatively cluster genes that are related to a specific biologic parameter. A number of methodologies have been employed for clustering, including hierarchical clustering as applied originally to microarray data sets by Eisen and colleagues,23 self-organizing maps (SOM) first applied to the distinction of AML and ALL by Golub et al,16 k-means, and principal component analysis (PCA). Unsupervised learning approaches have the advantage of being unbiased and allow for the identification of structure in large and complex data sets. However, because many different relationships or clusters may be possible in complex data sets, there may not be one “right” answer, a fact that is rarely appreciated by current readers and reviewers of the literature. Additionally, the clusters derived may not reflect important biological or clinical parameters.
In contrast to unsupervised learning tools, supervised learning methods, also termed “class predictors,” use various computational algorithms to model or “fit” a data set in order to be able to predict a specific label (such as AML vs ALL, remission vs failure, the presence of a specific cytogenetic abnormality vs not). In supervised learning, a data set is usually divided into a “training” set in which the parameter to be predicted is known to the analyst who then focuses on defining genes and profiles that can be used to predict for this parameter. Held in reserve, and blinded to the analyst, is a set of “test” cases in which the predictive profiles developed from the training set can be tested. One of the problems with supervised learning is that sample labels must be accurate and precise, which can be difficult in heterogeneous diseases. Another frequent problem with supervised learning approaches is that it is very easy to “over-fit” the data on a training set, resulting in an inability to make accurate predictions on the test set. Many supervised learning methods have been employed for the analysis of gene expression profiles, including Bayesian networks (P. Helman, R.Veroff, S. Atlas, C. L. Willman, unpublished data) and Support Vector Machines with Recursive Feature Elimination (SVM-RFE).27 These computational approaches require significant computational resources and parallel computing, particularly if predictive genes are to be rigorously validated. Many of these techniques, particularly when “leave one out” cross-validation (LOCV) is performed on large data sets, require extensive parametric studies or the solution of large matrix problems that can only be done using massively parallel computers. Ultimately, hypotheses generated from the computational analysis of an initial microarray data set must ideally be tested and validated on an independent sample set.
Discovery of Novel AML Classes by Using Gene Expression Profiling
Two recent studies have unexpectedly demonstrated that gene expression profiling can identify novel intrinsic biologic groups of AML patients that cannot be precisely defined by traditional morphologic, immunophenotypic, and cytogenetic classification schemes. While not surprising in the context of solid tumors, these results were indeed surprising in acute leukemia, in which we hold detailed knowledge of the cytogenetic and molecular genetic features of the disease. If validated, these studies hold promise to enhance and significantly alter our diagnostic and risk classification schemes for acute leukemia and to reveal unique clusters of cases that may benefit from specific therapeutic approaches.
Therapy-related AML
Gene expression profiles were recently obtained from enriched CD34+ AML progenitor cells from 14 t-AML patients using oligonucleotide arrays by Le Beau and colleagues.28,29 Unexpectedly, using hierarchical clustering, these investigators found 2 intrinsic groups of t-AML that would not have been predicted by clinical course, morphology, or cytogenetic abnormalities. The first group, containing t-AML patients with complex rearrangements involving chromosome 5q, had relative increases in expression of genes involved in cell cycle or checkpoint control (CCNA2, CCNE2, CDC2, and BUB1), cell growth (MYC), and loss of expression of the gene encoding interferon consensus sequence binding protein (ICSBP). The second group with a distinct gene expression profile clustered several t-AML patients with heterogeneous karyotypes including those with loss of chromosome 7, normal karyotypes, and t(3;3) or t(3;21). Despite cytogenetic heterogeneity, this group of patients had a similar gene expression profile with a relative decrease in expression of key transcriptional regulators (TAL1, GATA1, and EKLF) and overexpression of FLT3 and BCL2. These preliminary studies suggest that expression profiling may define novel biologic groups of t-AML not precisely related to karyotype that may benefit from a common therapeutic approach.
Infant AML/ALL
Our studies in infant leukemia have also yielded interesting and unexpected results. With its distinguishing genetic, biologic, and epidemiologic features, infant acute leukemia affords a unique investigational model for the study of leukemogenesis. Epidemiologic and molecular genetic studies have confirmed that most, if not all, infant leukemias arise in utero,30 suggesting a prenatal exposure or initiating event. AML and ALL also occur with relatively equal frequency in this age group. Although the mechanism is not well understood, structural rearrangements of the MLL/ALL1/HRX gene on chromosome 11q23 (hereafter referred to as MLL) appear to occur more frequently during fetal development, accounting for the relatively high incidence of abnormalities involving this gene in infant leukemia. Indeed, nearly 60% of infant leukemia cases of both ALL and AML morphologic subtypes have balanced, reciprocal translocations involving MLL fused to a number of different partner genes. With an overall survival of only 25%, there is an urgent need to better understand the etiology and pathogenesis of this disease in order to develop more effective therapies.
To improve diagnosis and risk classification and to identify new therapeutic targets in infant leukemia, we performed gene expression profiling using oligonucleotide arrays (Affymetrix U-95A.v2) in a retrospective cohort of 126 infant leukemias (78 ALL, 48 AML, 57/126 with MLL rearrangements) registered to NCI-sponsored clinical trials. To determine whether there were inherent biologic classes of infant leukemia, gene expression profiles were correlated with a large number of biologic and clinical covariables using several unsupervised clustering methods and novel data visualization tools (VxInsight) (Figure 6, see Color Figures, 513). For “class prediction” (AML vs ALL, MLL vs not, remission vs failure, the presence of specific cytogenetic abnormalities, and novel diagnostic and therapeutic targets), several supervised learning methods were employed (Bayesian networks, SVM-RFE, and Neuro-Fuzzy Logic).
VxInsight,31 a very powerful tool for class discovery and data visualization, was developed by our collaborator George Davidson and colleagues at Sandia National Laboratories in Albuquerque, NM (Figure 6, 513).28,32 VxInsight has the capacity to cluster patients or genes, using all of the gene expression data without having to select smaller subsets of genes for actual clustering, in a novel and intuitive way. Similar genes are clustered together spatially and represented in a 3-D terrain map, where large mountains represent large clusters of similar genes, and smaller hills represent clusters with fewer genes (Figure 6, 513). Clusters that are the most similar (genes or patients) are also sited nearer to each other and farther away from less similar clusters. The analogy to real terrain allows memorization of the landscape, which makes subsequent explorations very easy. The level of detail can be changed to “fly” over the terrain and view smaller clusters within clusters. The results of database queries can then be shown within the context of the clusters by highlighting those genes matching the queries. VxInsight also provides a simple way to select genes and display Web-based information for those genes. In addition, clinical and laboratory covariables can be loaded into the program, and the visual display and clusters can be queried in real time and reconfigured to show the interaction between the patients or genes in a cluster and these clinical and laboratory covariables.
When VxInsight31 was applied to our infant leukemia data, we discovered 3 discrete, statistically robust biologic clusters of infant leukemia. These intrinsic biologic groups could not be identified using traditional diagnostic parameters (morphology: ALL vs AML, immunophenotyping or cytogenetics: MLL rearrangement vs not) (Figure 7, see Color Figures, 513). We had anticipated that we might see 2 clusters (ALL vs AML or MLL vs not) or 3 clusters (ALL vs AML vs MLL leukemias) based on the data by Armstrong et al17 suggesting that MLL is a homogeneous and distinct form of leukemia. In each cluster, an individual patient is represented by a pyramid (highly similar patients in each cluster will be overlapping in this “high level” view) (Figure 7, 513). By querying VxInsight in “real-time,” cases previously labeled ALL vs AML were identified (ALL cases are colored white and AML colored green), and using analysis of variance (ANOVA), the most statistically significant genes that were unique to each cluster were identified (Figure 7, 513). These genes serve as potentially new diagnostic and therapeutic targets. One cluster (n = 52) (left, VX-GB, Figure 7, 513), predominantly lymphoid (51 ALL, 1 AML; 28 MLL), was uniquely characterized by expression of lymphoid, viral, and viral-induced genes. A second heterogeneous cluster (n = 54, 12 ALL, 42 AML; 16 MLL) (bottom right, VX-GC, Figure 7, 513) was associated with activation of DNA repair, metabolic, and RAS family genes, suggesting environmental initiating events. The third cluster (n = 21, 16 ALL, 5 AML; 10 MLL) (top, VX-GA, Figure 7, 513), with the highest rate of treatment failure, had a highly unique expression profile with activation of novel embryonal, neuronal, tumor suppressor, vascular, and adhesion genes, as well as genes associated with the earliest phases of hematopoiesis. We speculate that these “leukemic” cases, never before recognized or identified, represent transformed multipotential human stem cells. Each of the clusters we have identified may represent distinct etiologic pathways in infant leukemia in which MLL rearrangements act as a secondary, not a primary, transforming event. We hypothesize that identification of these novel groups may be important for future risk classification and therapeutic targeting in infants. Ongoing expression profiling studies of large retrospective cohorts of adult de novo and secondary AML cases are currently under way in our group and others.
Discovery of Gene Expression Profiles Associated with AML-Associated Cytogenetic Abnormalities: New Modes of Classification and Therapeutic Targets
Trisomy 8
Focusing on obtaining gene expression profiles associated with trisomy 8 and with normal cytogenetics in AML, Virtaneva et al21 used oligonucleotide arrays containing over 6000 genes to compare expression profiles in 10 AML samples with normal cytogenetics, 10 AML samples with trisomy 8 (+8), and bone marrow controls. While all AML samples had different gene expression profiles than normal CD34+ cells, it was difficult to discern differences in profiles between AML cases with +8 and those with normal cytogenetics. Cases with +8 clearly had increased expression of chromosome 8 associated genes, reflecting increased gene dosage and down-regulation of genes involved in the control of cell death.
Favorable cytogenetics: t(8;21), inv(16), t(15;17)
Schoch et al22 have used oligonucleotide arrays in 37 AML patients characterized by favorable cytogenetic abnormalities. A limited number of genes (n = 36) accurately separated AML patients with these different cytogenetic abnormalities. It remains to be determined whether an expansion of these results will lead to refinements in cytogenetic classification schemes. With the limited number of patients reported to date, the extent of heterogeneity in gene expression profiles in each cytogenetic abnormality is also unknown. Whether any of the genes identified are useful as new therapeutic targets remains to be determined.
MLL rearrangements
As discussed above, translocations involving the MLL gene at chromosome band 11q23 are the most common genetic alteration in both ALL and AML in infants and occur frequently in adults with both de novo and secondary AML (Table 9). While the t(4;11) is most common (occurring in 20%-30% of infant cases, primarily ALL), the t(9;11), t(1;11), t(11;19) in 2 variants, t(10;11), and many other translocation fusion partners may be seen (Table 9). While there is evidence that most of these chromosomal rearrangements carry a poor prognosis, the pathogenesis and unique gene expression profiles for each MLL translocation variant remain undefined. Using hierarchical clustering and PCA, Armstrong and colleagues17 concluded that leukemias with MLL rearrangements were a homogeneous and unique form of acute leukemia, distinct from AML and ALL. Several genes, including FLT3, were identified as being uniquely expressed in MLL-associated cases.17 However, these conclusions must be carefully considered in light of the cohort of MLL patients examined and their comparison AML and ALL groups. Of the 37 cases tested, 17 had MLL rearrangements, all but one of which was a t(4;11). The ALL comparison group contained a large number of cases with t(12;21). Thus, in this context, it is perhaps not surprising that MLL cases were deemed homogeneous and distinct from ALL cases with t(12;21) and other ALL and AML cases.
We recently used oligonucleotide arrays and high performance computing methods for both unsupervised and supervised data analysis to study MLL-associated gene expression profiles from the infant leukemia cohort, 54/126 of whom had heterogeneous MLL rearrangements: 30 t(4;11), 10 t(11;19), 6 t(10;11), 4 t(9;11), 4 t(1;11). As mentioned earlier, in the infant cohort, the presence or absence of MLL rearrangements did not define a distinct class during unsupervised learning approaches (Figure 7, 513). Using supervised learning algorithms for class prediction (Bayesian networks, SVM-RFE, and Discriminant Analysis), we found that MLL-associated gene expression profiles were quite heterogeneous among different MLL translocation variants. In addition, in the context of an appropriate control group of other infant cases, we found that the MLL genotype alone did not define a distinct group of leukemia cases, distinct from other ALL and AML cases arising in infants. Using these methods, we ordered the most statistically significant genes associated with those infant cases containing vs lacking any MLL rearrangement, which are therefore common to all MLL variants, as well as those genes that were unique to each translocation variant (Table 10 ). We believe that these genes represent novel diagnostic and therapeutic targets for MLL-associated disease. Although we do not agree with Armstrong et al17 that MLL cases are homogeneous, it is interesting that the lists of statistically significant genes that are shared by all MLL cases, particularly t(4;11), are virtually identical in our different laboratories. This result adds further support to the validity and reproducibility of this approach.
Discovery of Gene Expression Profiles that Predict for Treatment Failure in AML
Since Golub et al16 first identified genes, such as Hox A9, potentially associated with poorer outcome in AML, several groups have embarked on obtaining gene expression profiles that might predict for remission or failure in acute leukemia. Modeling of these complex clinical parameters is far more difficult than modeling other more uniform biologic and cytogenetic features. A second goal of our studies was to identify sets of genes that could be used to predict outcome as well as therapeutic resistance and treatment failure in AML and ALL. This is a more complex task, as the biologic or clinical labels for “continuous complete remission (CCR)” and “failure/relapse” are not pure or precisely defined and are dependent on time and numerous host and tumor factors. In addition, there is a tendency to “over-fit” algorithms and computational approaches to make these predictions, particularly in leukemia data sets where the failure rate to current therapies exceeds 50%. Nonetheless, identification of genes and pathways associated with treatment failure is essential to understanding therapeutic resistance and the development of appropriate therapies that overcome resistance.
In our studies, Bayesian nets and SVM-RFE have shown classification rates in excess of 90% in discerning between the type classes ALL vs AML (Table 11 ). However, only SVM-RFE was able to identify remission vs failure across the unconditioned data set with a total error rate differing from random prediction at a significance level of P < .10. Interestingly, far greater success is obtained when remission vs failure is conditioned on the VxInsight clusters (Figure 7, 513), rather than on AML vs ALL or in modeling the entire group as one biologic cohort. These results speak to the further biologic validity of the novel clusters that we have identified. In particular, significance levels of better than P = .01 were achieved within cluster VX-GC by the Bayesian nets (better than P = .05 for SVM), better than P = .10 within cluster VX-GA by both methods, and better than P = .175 within VX-GB by Bayesian nets. It is rather unlikely that random chance alone would produce such high accuracy levels. The results are taken as strong evidence that the VxInsight clusters reflect biologically real and clinically exploitable groupings of the patients. In contrast, comparable accuracy was not achieved conditioning on either of the traditional criteria of ALL versus AML or MLL versus not MLL.
Striking in VxInsight Group C (VX-GC, bottom right, Figure 7, 513), containing the majority of AML as well as a significant number of ALL cases, was the identification of genes and pathways associated with treatment failure. Interestingly, nearly 100 genes were determined to be significantly associated with failure, and there was very significant overlap in the list of genes derived from SVM-RFE and Bayesian approaches. The majority of the 20 most significant genes associated with treatment failure were those involved in specific signal transduction pathways (Table 11). We were struck by the appearance of a series of genes that encode signaling proteins (Table 11), all of which could potentially be placed in signaling cascades leading to RAS by both conventional and novel pathways. Other critical pathways involved DNA repair, apoptosis, and methylation.
Kinnunen et al33 have identified a statistically significant (P < .01) associated with relapse in pediatric AML. These include genes involved in proliferation (TTK S/T kinase, CRIP1, FTH1, BUB3, S100, and CA9), transcription (PRO2000, ZNF268, JUNB, MBLR, and EGR1), cellular trafficking (TRAM, RABEX5, MYO1B, and RANBP2L1), and oncogenesis (TTK, FOS, and CD68). The ability of these genes to predict for relapse and a poorer outcome are being tested in an independent set of cases.
Therapeutic Targets and Novel Approaches Derived from Gene Expression Profiling
Signal transduction targets
FLT3. One of the most interesting genes to emerge in the past 2 years as a diagnostic and therapeutic target in AML is FLT3. FLT3 is a transmembrane tyrosine kinase growth factor receptor (a member of the PDGF, FMS, and KIT receptor family) that is selectively expressed on hematopoietic cells, where it mediates stem cell proliferation and differentiation. Activation of the FLT3 receptor in AML precursor cells appears to stimulate proliferation and inhibit apoptosis. Several recent studies34–,39 have shown that an FLT3 mutation in AML (an in-frame internal tandem duplication of exon 11 and 12, termed the FLT3 ITD), results in auto-activation of the receptor in the absence of ligand, leading to independent activation of cell growth mediated by the RAS and STAT5 signaling pathways. Based on data recently published by several groups, FLT3 mutations are now considered one of the most common mutations in AML, increasing in frequency from 17% in pediatric AML35 to 36% in older AML patients.39 Although initially associated with a high WBC and normal karyotypes in AML, the FLT3 ITD has now been reported in many cytogenetic categories in both favorable and intermediate risk groups (Table 9).36 Although the prognostic significance of FLT3 ITD may be blunted by more intensive treatment regimens,37 multivariate analysis has shown that the FLT3 ITD is a striking predictor of poor outcome in pediatric and adult AML.35,36 In adults, FLT3 ITD was the most significant prognostic factor in predicting relapse risk and disease-free survival, as well as overall survival.36 The impact of FLT3 ITD may also be influenced by loss of the wild-type FLT3 allele; AML patients with FLT3 ITD and loss of the wild-type allele had the worst outcomes overall.38 With the development of selective tyrosine kinase inhibitors for FLT3-mediated signaling entering early phase clinical trials,40 there is a lot of excitement. Ultimately, careful consideration about how such selective inhibitors might be targeted with other agents will likely be essential for increased therapeutic outcomes in AML.
Many gene expression profiling studies have found relatively high levels of expression of presumably wild type FLT3 in AML, including AML and ALL cases associated with MLL rearrangements, cases of t-AML associated with abnormalities of chromosomes 3 and 7, and AML cases with t(15;17).17,22 However, as the status of the FLT3 gene was not determined in these samples, it remains possible that arrays could be detecting expression of both wild-type and mutant alleles. These data lead to speculation that FLT3 might be an attractive therapeutic target in these diseases, as well as in AML cases with FLT3 ITD. Very preliminary data (D. Small and J. Griffin, personal communications) have revealed that FLT3 inhibitors can inhibit signaling from overexpressed wild-type receptors as well as from mutant FLT3 receptors. If confirmed, the identification of high FLT3 expression in certain AML cases using arrays would be the first proof of principle that arrays can indeed identify novel therapeutic targets.
RAS. RAS mutations and RAS activation occur in a significant number of AML and MDS cases.6,41–,43 Newer therapies using farnesyl transferase inhibitors (FTI) (such as R115777; which target RAS to the plasma membrane, essential for RAS activation) or monoterpenes hold promise for therapeutic efficacy in AML.6,43 Interestingly, no correlation was observed between RAS mutations and response in early phase clinical trials testing these inhibitors,43 which suggests that RAS activation may be present in many patients regardless of the presence or absence of RAS mutations (as suggested in our array studies). Alternatively, FTIs may impact other key signaling elements in the RAS signaling pathways, such as Rho proteins or PI3 Kinase/AKT, which promotes cell survival. Our recent studies in pediatric leukemia suggest that perturbations in RAS signaling pathways are common in AML as well as in related cases of ALL (Figure 7, 513). In many AML cases, RAS may be activated by perturbations in upstream signaling pathways (such as FLT3, KIT, or PDGF).
Intrinsic drug resistance, apoptosis, angiogenesis, and adhesion. Each of these biologic features of leukemic cells may mediate drug resistance and promote cell survival. Each pathway also represents an important potential avenue for new drug development and therapeutic targeting.12,13,44– 46 It is hoped that as more gene expression profiling data are compiled, we can determine how these pathways are affected in each type of AML in relation to existing cytogenetic and other prognostic abnormalities. This knowledge may allow us to appropriately target groups of patients to more effective experimental treatment approaches.
Summary and Future Scientific Questions
Exciting preliminary gene expression profiling studies are providing new insights into the etiology and pathogenesis of AML. These studies hold promise to impact disease classification, risk assessment, and therapeutic targeting and will likely lead to the identification of novel therapeutic targets. To make continued progress in these studies, it will be essential to obtain gene expression profiles on large, statistically designed cohorts of well-characterized leukemic patients for whom detailed cytogenetics, biologic covariables, and clinical outcome parameters are known and available. Particularly fruitful areas for future investigation include the application of genomic and proteomic technologies to understand alterations in the global patterns of gene expression and protein function in AML, the development of computational methods to place genes into pathways in order to determine the impact of aggregate expression profiles on cell behavior, the development of computational methods to prioritize target selection for drug development, and the development of common database architectures so that data may be exchanged and shared to facilitate progress in this field.
IV. New Agents and Strategies in Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndromes
Francis J. Giles, MD,*
M.D. Anderson Cancer Center, Department of Leukemia, 1400 Holcombe Blvd., Box 428, Houston, TX 77030
With current standard induction regimens for adult patients with AML, the CR rates range from 40% to 90%, and the cure rates from less than 10% to 70%, depending on several characteristics, including leukemia karyotype, patient age and performance status, and organ function (Table 12 ).1–,4 Patients with high-risk myelodysplastic syndrome (MDS), defined here as MDS with 10% or more marrow blasts, have a prognosis similar to that of patients with AML.5–,7 High-risk MDS and AML have different pathophysiologies (e.g., rates of apoptosis, methylation profiles, incidence of FLT-3 internal tandem duplications [ITD] or mutations, aberrant angiogenesis, angiogenic factor profiles).8–,18 Some anti-AML programs have been applied successfully to high-risk MDS,1,6,19–,21 while other approaches are being designed to specifically target the pathophysiologic events in MDS.22– 25 Herein we will review some of the novel agents under investigation by our group in adult patients with AML or high-risk MDS.
Special Therapeutic Considerations in AML with Favorable Karyotypes
These include t(8;21), inversion 16, and APL, i.e., t(15;17) and variants. In AML with t(8;21), multiple high-dose ara-C regimens have improved outcome. In a review of CALGB studies, 29 patients with t(8;21) received 3 or more cycles of high-dose ara-C and 21 patients received only 1 cycle. The results, shown in Table 13 , indicated significantly lower relapse rates, better 5-year survival, and better overall disease-free survival (DFS) rates with more high-dose ara-C consolidations.26 Inversion 16 AML also benefits from high-dose ara-C in relation to decreased incidence of central nervous system disease (from 30% to less than 5%). However, the importance of multiple high-dose ara-C courses, although possibly as significant as with t(8;21), has not been analyzed.
The introduction of ATRA to APL therapy has improved prognosis. ATRA plus chemotherapy has improved the cure rate from less than 40% to over 70%.27–,31 Anthracyclines are very important for improved outcome,29,32 while ara-C plays little if any role.30,33 Polymerase chain reaction (PCR) monitoring for promyelocytic leukemia gene-retinoic acid receptor alpha (PML-RARα) gene is part of the management of patients on therapy in first CR, as molecular relapses can still be cured in 70% of cases.34–,36 Arsenic trioxide (As2O3) is important for salvage therapy and is probably superior to ATRA.36 The updated results of the pivotal trial of As2O3 in APL salvage included 52 patients, 45 of whom achieved CR (87%). Molecular CR was noted in 51% of patients at CR and in 78% after 1 consolidation course. Persistent DFS was observed in 1 of 7 patients (14%) who received only 1 course of As2O3, in 8 of 20 patients (40%) who received 4 or more courses of AS2O3 with or without other therapies, and in 11 of 12 patients (92%) who underwent allogeneic stem cell transplant (SCT) in CR.
Studies of As2O3 plus ATRA in frontline therapy are ongoing and may obviate the need for any chemotherapy. Gemtuzumab ozogamicin (GO; Mylotarg) is also effective against APL (strong CD33 expression) and is currently being investigated in combination with ATRA in frontline and salvage APL.37 In a recent M.D. Anderson Cancer Center (MDACC) frontline study of ATRA + GO in APL, patients received induction with ATRA 45 mg/m2 daily (D) until CR, then for 2 weeks every 4 weeks for 9 months, and GO 9 mg/m2 intravenously (IV) on day 1 for induction, then every month. Idarubicin 12 mg/m2/D × 3 was given during induction for high-risk patients (peripheral blasts more than 10 × 109/L) or if PCR persisted as positive or reverted to positive after 3 months into CR. Among 20 patients treated, 16 achieved CR including 14 of 17 low-risk and 2 of 3 high-risk patients. After a median follow-up of 9 months, all 16 patients remain in CR; only 1 became transiently positive. This experience suggests that future regimens in APL need not incorporate ara-C, and, in most patients, may not require anthracyclines, and that long-term event-free survival (EFS) would be obtained with nonchemotherapy regimens (e.g., ATRA plus As2O3) or without anthracyclines (e.g., ATRA plus GO). Novel approaches that are being investigated as induction and/or maintenance/consolidation approaches include liposomal ATRA, Am80, GO, HuM195, and tetra-arsenic tetra-sulfide.37–,42 The role of SCT in patients with APL is unclear, with most reports based on registry data or small patient series.43 The severe, often fatal, coagulopathy that is often present in high-risk APL patients remains a serious challenge.44
Strategies to Improve Prognosis of AML and High-Risk MDS
Current promising investigational avenues in AML-MDS include both traditional cytotoxic approaches and novel “targeted” or immunomodulatory therapies.
Intensification strategies, e.g., dexamethasone, cytarabine, thioguanine, etoposide, and daunorubicin (DCTER) regimen as used in pediatric AML.45,46
Investigating new formulations of established drugs (e.g., liposomal formulations of daunorubicin,47,48 asparaginase,49 pegylated interferon,49 topoisomerase I inhibitors,50 deposomal ara-C for intrathecal therapy51).
Investigating drugs with mechanisms of actions not fully exploited in current AML-MDS therapies. Examples include:
Strategies directed at relatively leukemia-specific mechanisms or antigens, including:
Exploring improved formulations of established agents that offer pharmacologic or safety advantages
A recent example of such strategies is the development of liposomal daunorubicin.47,48 We initially studied liposomal daunorubicin in a Phase I study as a single agent at doses of 75, 100, 150, and 200 mg/m2/D × 3 (225 to 600 mg/m2 per course) in patients with refractory leukemia.48 Among 24 patients studied, the maximum tolerated dose (MTD) was defined at 150 mg/m2/D × 3, and the dose limiting toxicity (DLT) was mucositis. No cardiac events were noted; 2 patients achieved CR. These data led to the next study, a Phase I-II study of liposomal daunorubicin 75 to 150 mg/m2/D × 3 plus ara-C 1 g/m2 by continuous infusion daily × 4 in 62 patients.47 Overall 18 patients (29%) achieved CR and 7 marrow CR with low platelets (CRp, 11%), for an overall response rate of 40%. The observed:expected CR ratio was 1.7, suggesting a beneficial effect. The median CR duration was 14 months, and median survival 6 months. Mucositis was DLT occurring in 4 of 9 patients at 150 mg/m2/D × 3 but in only 2 of 32 (6%) at 125 mg/m2 and in 1 of 13 (8%) at 135 mg/m2. Severe cardiotoxicity was noted in 4 patients (6%). Liposomal daunorubicin combination regimens are being investigated as frontline therapy in AML and ALL, at doses of 125 mg/m2/D × 3, by several groups.
Investigating agents with different mechanisms of action
Combinations of topotecan + ara-C and fludarabine + ara-C. These regimens have been extensively used in salvage and frontline therapy of AML, MDS, and ALL.1,6,21,65 Topotecan + ara-C has high activity and relatively low toxicity in advanced MDS.66 Among 59 patients with MDS and 27 with chronic myelomonocytic leukemia (CMML) treated with topotecan 1.25 mg/m2 continuous infusion (CI)/D × 5 and ara-C 1 g/m2 over 2 h/D x 5, the CR rate was 56% and the induction death rate 7%. These regimens are probably equivalent to idarubicin + high-dose ara-C (IA) in AML and high-risk MDS and could be used as alternative or complementary strategies (Table 14 ).1,6 New topoisomerase I inhibitors under study include liposomal lurtotecan (OSI 211),50 DX-8951f,67,68 and pegylated preparations (GL147211C).69
Nucleoside analogues. Clofarabine is an adenosine nucleoside analog that was designed to combine the better properties of fludarabine and chlorodeoxyadenoside (Figure 8 ).54,55 Phase I studies of clofarabine 2-55 mg/m2/D × 5 established the phase II dose at 40 mg/m2/D × 5; the DLTs were liver dysfunction and skin rashes. Responses were noted in 6 of 35 patients (17%). In the ongoing Phase II study, 39 patients have been treated. Responses in the 36 evaluable patients with refractory-relapse leukemias were encouraging (Table 15 ). Clofarabine is currently undergoing multi-institutional studies in pediatric and adult leukemias. Combination studies will include clofarabine + ara-C ± idarubicin.
Troxacitabine is an L isomer cytosine analogue that has significant activity in AML and CML blastic phase (Figure 8).56,70 It has a number of metabolic features that may allow activity in ara-C resistant AML.71–,73 Deoxycytidine kinase (dCK), which lacks chiral specificity, catalyses the monophosphorylation of both ara-C and troxacitabine. Deoxycytidine deaminase (dCD) is more chiral specific and cannot inactivate troxacitabine. Troxacitabine has a unique pattern of cellular uptake and metabolism, which may also allow it to circumvent another mechanism of resistance to cytotoxic nucleoside analogs. Nucleoside-specific membrane transporters (NSMT) mediate plasma membrane permeation of many nucleoside analogs including ara-C and fludarabine. In some preclinical models, troxacitabine is transported rapidly into cells by both equilibrative sensitive and insensitive nucleoside transport systems with subsequent accumulation of troxacitabine monophosphate, diphosphate, and triphosphate in a time- and concentration-dependent manner. As troxacitabine is not dependent on NSMT to achieve a lethal intracellular concentration, it may thus not be susceptible to NSMT-mediated mechanisms of resistance to ara-C. Unlike ara-C, which lacks proportionality between ara-C TP formation and extracellular ara-C concentration, the formation of troxacitabine diphosphate increases linearly with increasing extracellular drug concentration. Troxacitabine does not inhibit ribonucleotide reductase and may thus be mechanistically complementary to several nucleosides that are cytotoxic in part via ribonucleotide reductase inhibition e.g., Gemcitabine, FMdC, fludarabine. The pharmacokinetic behavior of troxacitabine is substantially different from that of other nucleoside analogs possessing a D configuration, which are characterized by rapid disappearance from plasma due to deamination. In contrast, troxacitabine has a long terminal half-life (82 hours) and a systemic clearance comparable to the glomerular filtration rate (137 mL/min).56
In a Phase I study, doses ranging from 0.72 to 10 mg/m2 IV over 1 hour daily × 5 were investigated in patients with refractory leukemia.56 Response was as follows: AML, 3 CR + 1 PR in 31 patients (13%); MDS, 1 marrow CR in 6 patients (16%); CML blastic phase, return to chronic phase in one patient. The study defined the MTD of troxacitabine at 8 mg/m2/D × 5; DLTs were skin rashes and mucositis. Phase II studies of troxacitabine alone and in combination have been completed in AML and led to selection of the combinations of troxacitabine + ara-C and troxacitabine + idarubicin for further testing in frontline AML.70 An MDACC frontline study comparing IA versus troxacitabine + ara-C versus troxacitabine + idarubicin in an adaptive randomization design, with CR by day 50 as the primary efficacy endpoint, showed IA to be superior to both investigational arms in elderly patients with an adverse karyotype. Troxacitabine is being investigated in a multicenter randomized study in patients with refractory AML and merits further study in patients with CML blastic phase.
GO (Mylotarg, CMA-676)
GO has been approved by the Food and Drug Administration for the treatment of patients 60 years or older in first relapse with first CR durations of 3 to 6 months or more and who are considered unsuitable for standard cytotoxic therapy.74 Approval was based on data from 3 Phase II pivotal trials in 142 patients in which GO induced a CR rate of 15%.75
GO has been tested at 9 mg/m2/D and 8 or 15 ± interleukin-11 (IL-11) (20 μg/kg/D) in 54 patients with AML-MDS (> 10% blasts) at high risk of chemotherapy-related mortality (age ≥ 65 years; induction mortality 30%-40%).76 The median age was 72 years (range 65-89 years). Results are shown in Table 16 . We concluded that GO alone is less active than chemotherapy in frontline therapy of elderly patients with AML-high risk MDS. IL-11 may have direct undefined anti-AML efficacy. GO has now been combined in several studies, with IA, fludarabine + ara-C, topotecan + ara-C, and IL-11.76 A significant challenge with GO therapy, particularly where it is combined with other cytotoxic agents, is hepatic venoocclusive disease (VOD).77–,80 The incidence of VOD may be related to the dose of GO used and may be less when GO is used in the maintenance setting or in APL.40
Farnesyl transferase inhibitors
Several farnesyl transferase inhibitors (FTI) are under investigation including R115777, which has shown activity in AML, Ph-positive CML, CMML, MDS, and myelofibrosis; Schering 66336, which has shown activity in CMML and MDS; and BMS 214662.
In a Phase I study, R115777 was given orally at doses of 100 to 1200 mg twice daily × 4 weeks every 6 weeks.81 It induced 2 CR + 6 PR (32%) in 25 patients with AML relapse. The MTD was 300-600 mg orally twice daily and was dose-limited by central nervous system and peripheral neurotoxicity and fatigue. In Phase I-II studies, R115777 was given to 44 patients with MDS at 300 to 600 mg twice daily × 3 to 4 weeks every 4 to 6 weeks. It induced responses in 8 of 34 (24%) evaluable patients. Ongoing studies are further exploring the activities of R115777 and Schering 66336 alone or in combinations in chronic myeloid leukemia (CML) (with imatinib), MDS, and myelofibrosis.
MDR modulation
In a Southwestern Oncology Group (SWOG) study in refractory-relapse AML, 226 patients received daunorubicin 45 mg/m2/D × 3 (days 6-8) + ara-C 3 g/m2/D × 5 (days 1-5); half were randomized to cyclosporin 16 mg/kg/D by continuous infusion on days 6-8.58 Results were significantly better in the cyclosporin arm (Table 17 ). Studies of the less immunosuppressive MDR modulator, PSC 833, are ongoing in both the salvage and de novo AML settings.
Targeting angiogenesis
Increased marrow vascularity and increased levels of plasma-cellular vascular endothelial growth factor (VEGF) and other angiogenesis modulators have been demonstrated in AML, MDS, CML, and other leukemias and have been associated with poor prognosis in AML and CML.82 Thus, targeting angiogenesis may improve outcome. Thalidomide, a putative angiogenesis inhibitor, has shown activity in MDS, particularly in RA.23,83,84 A study at MDACC of liposomal daunorubicin + high-dose ara-C ± thalidomide did not show an added benefit with thalidomide. Other angiogenesis inhibitors (SU5416, SU11248, PTK787, GFKI-258, CPE-7055, VEGF MoAb) are undergoing investigation.11,64,82,85,86 Both in vitro direct inhibition of AML blast growth in response to cKit inhibition by SU5416, and clinical responses on a Phase II study have been reported in patients with AML.87,88 The assessment of these agents in Phase I studies (in which there may not be a traditional DLT) or in Phase II studies with statistics designed around expectations of achievement of objective responses, which in turn are based on our experience with standard cytotoxic agents, may not be optimal. Cytostatic agents may well be of most benefit in AML as a maintenance agent or in relatively indolent phases of MDS. Predictive methods to identify potential responders to angiogenic modulators are required.
Targeting methylation
DNA methylation is associated with suppression of regulatory genes and with disease progression.63 Azacytidine (AZA) and decitabine are active hypomethylating agents in MDS. Decitabine is also active in AML and CML.55,89,90 A randomized study of AZA versus supportive care showed a benefit from AZA, as measured by response rate, time to transformation, and quality of life (Table 18 ).91,92 Decitabine given at 45 mg/m2/D × 3 to 121 patients with MDS induced CR in 20%, PR in 10%, and improvement in 19%. Induction death was 8%. Response rates by IPSS risk were 58% in high-risk, 45% in intermediate-2, and 39% in intermediate-1. A Phase I low-dose longer exposure biologic study of decitabine defined the Phase II dose at 10 mg/m2/D × 10. Among 42 evaluable patients, 7 (17%) achieved CR, 10 (24%) had PR, and 7 (17%) improved. Combinations of decitabine (with ara-C in AML, with imatinib in CML, with topotecan or ara-C in MDS) are under consideration.
Other targets
Mutations and ITD of FLT-3 in AML are common (30%-40%) and associated with poor prognosis.4,14–,16,61 Several studies are now investigating FLT-3 inhibitors in Phase I-II studies of AML with FLT-3 mutation/ITD.
Overexpression of bcl-2, an anti-apoptosis signal, may be involved in AML resistance to therapy. Genasense (bcl-2 antisense) is undergoing evaluation in AML.55
A novel diphtheria toxingranulocyte-macrophage colony-stimulating factor, fusion protein (DT388 GMCSF)has completed Phase I evaluation in refractory-relapse AML.93 The drug was given as a short infusion daily × 5. The MTD was 4 μg/kg/D and DLT was liver toxicity. Among 31 patients treated, 1 had CR and 2 had PR (10%).
Conclusions
The plethora of available novel agents engenders hope that real progress will soon be made in achieving cure in all patients with AML or MDS. Above we have focused on some aspects of our investigational programsspace does not allow a discussion of other very important approaches including immunomodulation, vaccination, and SCT. A challenge with the increasing number of available agents and their diverse mechanisms of anti-leukemic activity is the rational design and conduct of studiesrecent advances in biostatistics should aid clinical research.94– 96 The top priority is to ensure access for patients to adequately designed clinical studies.
Extracellular matrix constituents.
| Proteoglycans and constituent glycosaminoglycans |
| Heparan sulfate |
| Chondroitin sulfate |
| Dermatan sulfate |
| Hyaluronic acid |
| Collagen: types I, III, IV, V, VI |
| Fibronectin |
| Thrombospondin |
| Sialoadhesin |
| Laminin |
| Tenascin |
| Proteoglycans and constituent glycosaminoglycans |
| Heparan sulfate |
| Chondroitin sulfate |
| Dermatan sulfate |
| Hyaluronic acid |
| Collagen: types I, III, IV, V, VI |
| Fibronectin |
| Thrombospondin |
| Sialoadhesin |
| Laminin |
| Tenascin |
Factors constitutively or inducibly expressed by marrow stromal cells.
| Abbreviations: G-CSF, granulocyte colony-stimulating factor; IL, interleukin; GM-CSF, granulocyte-macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; SCF, stem cell factor; LIF, leukemia inhibitory factor; TNF, tumor necrosis factor; TGF, tumor growth factor; HGF, hepatocyte growth factor; NGF, nerve growth factor; BDNF, brain-derived neurotopic factor; SDF-1, stromal-derived factor-1. | |
| G-CSF | IL-1 |
| GM-CSF | IL-1β |
| M-CSF | IL-6 |
| Flt-3 ligand | IL-7 |
| SCF | IL-8 |
| LIF | IL-12 |
| Thrombopoietin | IL-14 |
| TNF-α | IL-15 |
| TNF-β | IL-16 |
| TGF-β | IL-17 |
| HGF | IL-18 |
| NGF | |
| BDNF | |
| SDF-1 | |
| Abbreviations: G-CSF, granulocyte colony-stimulating factor; IL, interleukin; GM-CSF, granulocyte-macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; SCF, stem cell factor; LIF, leukemia inhibitory factor; TNF, tumor necrosis factor; TGF, tumor growth factor; HGF, hepatocyte growth factor; NGF, nerve growth factor; BDNF, brain-derived neurotopic factor; SDF-1, stromal-derived factor-1. | |
| G-CSF | IL-1 |
| GM-CSF | IL-1β |
| M-CSF | IL-6 |
| Flt-3 ligand | IL-7 |
| SCF | IL-8 |
| LIF | IL-12 |
| Thrombopoietin | IL-14 |
| TNF-α | IL-15 |
| TNF-β | IL-16 |
| TGF-β | IL-17 |
| HGF | IL-18 |
| NGF | |
| BDNF | |
| SDF-1 | |
Possible interactions between leukemic and stromal cells.
| Adapted from Dührsen and Hossfeld.11 |
| • Promotion of leukemic cell growth |
| Inhibition of apoptosis |
| Blockade of differentiation |
| Stimulation of proliferation |
| Growth stimulation by hepatocyte growth factors (HGFs) |
| • Inhibition of leukemic cell growth |
| Induction of differentiation |
| Inhibition of proliferation |
| • Inhibition of stromal cell growth |
| Adapted from Dührsen and Hossfeld.11 |
| • Promotion of leukemic cell growth |
| Inhibition of apoptosis |
| Blockade of differentiation |
| Stimulation of proliferation |
| Growth stimulation by hepatocyte growth factors (HGFs) |
| • Inhibition of leukemic cell growth |
| Induction of differentiation |
| Inhibition of proliferation |
| • Inhibition of stromal cell growth |
Adhesion molecule interactions.
| Adhesion Molecule | Location | Ligand | Location |
|---|---|---|---|
| Abbreviations: VLA, very late antigen; HP, hematopoietic progenitors; AML, acute myeloid leukemia; VCAM, vascular adhesion molecule; ECM, extracellular matrix; MSC, marrow stromal cells; LFA, lymphocyte function antigen; DHC, differentiated hematopoietic cells; ICAM, intercellular adhesion molecule; EC, endothelial cells. | |||
| β1 integrins | |||
| α4β1 (VLA-4) | HP | Fibronectin (CS1 domain) | ECM |
| AML cells | VCAM-1 | MSC | |
| α5β1 (VLA-5) | HP | Fibronectin (RGD sequence) | ECM |
| β2 integrins | |||
| LFA-1 | HP, AML cells | ICAM-1 | MSC, EC |
| Mac-1 | DHC, AML cells | ||
| Selectins | |||
| E-selectin | EC | Sialylated sugar moieties; Lewis x; CD44 | Myeloid cells, T cells |
| P-selectin | EC, platelets | P selectin, glycoprotein ligand 1 (CD162) | Myeloid cells, AML blasts |
| L-selectin | HP | Glycoprotein ligand | HP, AML blasts |
| CD44 | HP | Hyaluronic acid | ECM |
| AML blasts | Fibronectin | ||
| Adhesion Molecule | Location | Ligand | Location |
|---|---|---|---|
| Abbreviations: VLA, very late antigen; HP, hematopoietic progenitors; AML, acute myeloid leukemia; VCAM, vascular adhesion molecule; ECM, extracellular matrix; MSC, marrow stromal cells; LFA, lymphocyte function antigen; DHC, differentiated hematopoietic cells; ICAM, intercellular adhesion molecule; EC, endothelial cells. | |||
| β1 integrins | |||
| α4β1 (VLA-4) | HP | Fibronectin (CS1 domain) | ECM |
| AML cells | VCAM-1 | MSC | |
| α5β1 (VLA-5) | HP | Fibronectin (RGD sequence) | ECM |
| β2 integrins | |||
| LFA-1 | HP, AML cells | ICAM-1 | MSC, EC |
| Mac-1 | DHC, AML cells | ||
| Selectins | |||
| E-selectin | EC | Sialylated sugar moieties; Lewis x; CD44 | Myeloid cells, T cells |
| P-selectin | EC, platelets | P selectin, glycoprotein ligand 1 (CD162) | Myeloid cells, AML blasts |
| L-selectin | HP | Glycoprotein ligand | HP, AML blasts |
| CD44 | HP | Hyaluronic acid | ECM |
| AML blasts | Fibronectin | ||
Risk group definition used by transplant trial groups.
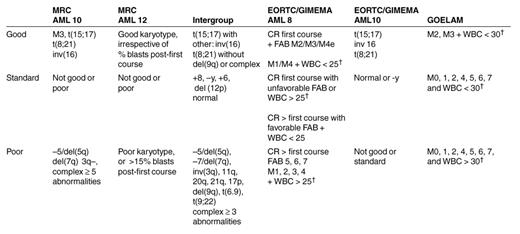
Abbreviations: MRC, Medical Research Council; AML, acute myeloid leukemia; EORTC/GIMEMA, European Organization for Research and Treatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; inv, inversion; del, deletion; CR, complete remission; FAB, French-American-British classification of acute leukemias; M, myeloid leukemia subtype; WBC, white blood cells.
†Number of WBC × 109/L.
Major randomized prospective trials.
| Intent to Treat/Actually Treated + Regimen | ||||||||
|---|---|---|---|---|---|---|---|---|
| Study Group, Years, Age range | No. of Patients Attained CR/ Total Patients | Induction | Postremission Therapy Before Receiving the Assigned Therapy (IC/ASCT/Allo SCT) | Randomized, % | Time of Randomization | Allo SCT | Assigned IC | ASCT |
| Abbreviations: CR, complete remission; IC, intensive chemotherapy; ASCT, autologous stem cell transplantation; allo SCT, allogeneic stem cell transplantation; EORTC/GIMEMA, European Organization for Research and Treatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; AML, acute myeloid leukemia; ABMT, autologous bone marrow transplantation; PBSCT, peripheral blood stem cell transplantation; BM, bone marrow; PB, peripheral blood; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; MRC, Medical Research Council; DAT, daunorubicin, cytarabine, thioguanine; ADE, cytarabine, daunorubicin, etoposide; MACE, amsacrine, cytarabine, etoposide; MIDAC, mitoxantrone, cytarabine; CTX, cyclophosphamide; TBI, total body irradiation; MAE, mitoxantrone, cytarabine, etoposide; S-DAT, daunorubicin, cytarabine 100/m2/d, thioguanine; H-DAT, daunorubicin, cytarabine 200/m2/d, thioguanine; Bu, busulfan. | ||||||||
| † Second course has been given to patients who failed to achieve CR with first induction. | ||||||||
| ‡1966 patients entered the study, but some were taken out because of incorrect diagnosis; 83% of the patients with confirmed AML attained CR. | ||||||||
| EORTC/GIMEMA AML 8 1986–1991 10-59 y | 623/941 (66%) | Daunorubicin 45 mg/m2 + cytarabine 200 mg/m2 1-2 courses† | Cytarabine 6 g/m2/course + amsacrine | 63 | After 2 (or 3) courses† | 144/168 (86%) | 104/126 (83%) cytarabine 16 g/m2/course + daunorubicin | 95/128 (74%) |
| EORTC/GIMEMA AML 10 11/1993–12/1999 16-60 y | 1445/2038 (71%) | Anthracycline + cytarabine + etoposide | Anthracycline + cytarabine | 67 | ABMT vs PBSCT | 198/292 (68%) | No arm for only IC | 87/146-BM 99/146-PB |
| GOELAM 11/1993–12/1999 15-50 y | 367/517 (71%) (87%)2 | Cytarabine 200 mg/m2 + anthracycline (1-2 courses)† | Cytarabine 500 mg/m2/course + amsacrine for pt assigned allo SCT cytarabine 24 g/m2/course + anthracycline for pt assigned ASCT/IC | 61 or | After 2 (or 3) | 73/88 courses† | 71/78 (91%) (83%) | 75/86 amsacrine + etoposide |
| MRC AML 10 1988–1966 < 55 y | 1609/1966‡ (83%) (66%) | DAT or ADE (1-2 courses)† | All patients received 3 additional courses of DAT/ADE (1), MACE (1), MIDAC (1) if they had not received 2 courses already | 34 | After 3rd | 257/419 course | No additional (61%) | 126/190 (4 courses previously) |
| MRC AML12 1995–2002 < 60 y | n = 3459 (85% CR) | 2 courses of ADE vs MAE replaced by S-DAT vs H-DAT | MACE (1) | After 3rd course | Still un-published | Randomization between 1 and 2 additional courses: ICE vs | Still un-published ICE followed by MIDAC | |
| Intergroup study 1990–1995 16-55 y | 518/740 (70%) | Cytarabine 100 mg/m2 + idarubicin | All patients who achieved CR received 1 course of cytarabine 500 mg/m2/course + idarubicin | 60 | After 2 (or 3) courses† | 92/113 (81%) | 106/117 (91%) cytarabine 36 g/m2/course | 63/116 (54%) |
| Intent to Treat/Actually Treated + Regimen | ||||||||
|---|---|---|---|---|---|---|---|---|
| Study Group, Years, Age range | No. of Patients Attained CR/ Total Patients | Induction | Postremission Therapy Before Receiving the Assigned Therapy (IC/ASCT/Allo SCT) | Randomized, % | Time of Randomization | Allo SCT | Assigned IC | ASCT |
| Abbreviations: CR, complete remission; IC, intensive chemotherapy; ASCT, autologous stem cell transplantation; allo SCT, allogeneic stem cell transplantation; EORTC/GIMEMA, European Organization for Research and Treatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; AML, acute myeloid leukemia; ABMT, autologous bone marrow transplantation; PBSCT, peripheral blood stem cell transplantation; BM, bone marrow; PB, peripheral blood; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; MRC, Medical Research Council; DAT, daunorubicin, cytarabine, thioguanine; ADE, cytarabine, daunorubicin, etoposide; MACE, amsacrine, cytarabine, etoposide; MIDAC, mitoxantrone, cytarabine; CTX, cyclophosphamide; TBI, total body irradiation; MAE, mitoxantrone, cytarabine, etoposide; S-DAT, daunorubicin, cytarabine 100/m2/d, thioguanine; H-DAT, daunorubicin, cytarabine 200/m2/d, thioguanine; Bu, busulfan. | ||||||||
| † Second course has been given to patients who failed to achieve CR with first induction. | ||||||||
| ‡1966 patients entered the study, but some were taken out because of incorrect diagnosis; 83% of the patients with confirmed AML attained CR. | ||||||||
| EORTC/GIMEMA AML 8 1986–1991 10-59 y | 623/941 (66%) | Daunorubicin 45 mg/m2 + cytarabine 200 mg/m2 1-2 courses† | Cytarabine 6 g/m2/course + amsacrine | 63 | After 2 (or 3) courses† | 144/168 (86%) | 104/126 (83%) cytarabine 16 g/m2/course + daunorubicin | 95/128 (74%) |
| EORTC/GIMEMA AML 10 11/1993–12/1999 16-60 y | 1445/2038 (71%) | Anthracycline + cytarabine + etoposide | Anthracycline + cytarabine | 67 | ABMT vs PBSCT | 198/292 (68%) | No arm for only IC | 87/146-BM 99/146-PB |
| GOELAM 11/1993–12/1999 15-50 y | 367/517 (71%) (87%)2 | Cytarabine 200 mg/m2 + anthracycline (1-2 courses)† | Cytarabine 500 mg/m2/course + amsacrine for pt assigned allo SCT cytarabine 24 g/m2/course + anthracycline for pt assigned ASCT/IC | 61 or | After 2 (or 3) | 73/88 courses† | 71/78 (91%) (83%) | 75/86 amsacrine + etoposide |
| MRC AML 10 1988–1966 < 55 y | 1609/1966‡ (83%) (66%) | DAT or ADE (1-2 courses)† | All patients received 3 additional courses of DAT/ADE (1), MACE (1), MIDAC (1) if they had not received 2 courses already | 34 | After 3rd | 257/419 course | No additional (61%) | 126/190 (4 courses previously) |
| MRC AML12 1995–2002 < 60 y | n = 3459 (85% CR) | 2 courses of ADE vs MAE replaced by S-DAT vs H-DAT | MACE (1) | After 3rd course | Still un-published | Randomization between 1 and 2 additional courses: ICE vs | Still un-published ICE followed by MIDAC | |
| Intergroup study 1990–1995 16-55 y | 518/740 (70%) | Cytarabine 100 mg/m2 + idarubicin | All patients who achieved CR received 1 course of cytarabine 500 mg/m2/course + idarubicin | 60 | After 2 (or 3) courses† | 92/113 (81%) | 106/117 (91%) cytarabine 36 g/m2/course | 63/116 (54%) |
Recent trials evaluating allogeneic transplant on a donor versus no donor basis.¶
| Disease-Free Survival* (%) | Overall Survival* (%) | |||
|---|---|---|---|---|
| Trial | Donor | No Donor | Donor | No Donor |
| Abbreviations: EORTC/GIMEMA, European Organization for Research and Treatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; AML, acute myeloid leukemia; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; MRC, Medical Research Council. | ||||
| ¶ All patients having a donor available were regarded as transplant recipients. | ||||
| * % at 4 years or beyond | ||||
| †P = .01 ‡ P = .046 | ||||
| EORTC/GIMEMA AML 8 | 46 | 33† | 48 | 40 |
| GOELAM | 44 | 38 | 53 | 53 |
| MRC AML 10 | 50 | 42 | 55 | 50 |
| Intergroup | 43 | 35 | 46 | 52 |
| EORTC/GIMEMA AML 10 | 51.4 | 41.2‡ | 58 | 49.4 |
| Disease-Free Survival* (%) | Overall Survival* (%) | |||
|---|---|---|---|---|
| Trial | Donor | No Donor | Donor | No Donor |
| Abbreviations: EORTC/GIMEMA, European Organization for Research and Treatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; AML, acute myeloid leukemia; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; MRC, Medical Research Council. | ||||
| ¶ All patients having a donor available were regarded as transplant recipients. | ||||
| * % at 4 years or beyond | ||||
| †P = .01 ‡ P = .046 | ||||
| EORTC/GIMEMA AML 8 | 46 | 33† | 48 | 40 |
| GOELAM | 44 | 38 | 53 | 53 |
| MRC AML 10 | 50 | 42 | 55 | 50 |
| Intergroup | 43 | 35 | 46 | 52 |
| EORTC/GIMEMA AML 10 | 51.4 | 41.2‡ | 58 | 49.4 |
Prospective trials of autologous transplantation in adults.
| Relapse* (%) | Disease-Free Survival* (%) | Overall Survival* (%) | ||||
|---|---|---|---|---|---|---|
| Trial | Autograft | Chemo | Autograft | Chemo | Autograft | Chemo |
| Abbreviations: EORTC/GIMEMA, European Organization for Research and Treatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; AML, acute myeloid leukemia; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; NA, not applicable; MRC, Medical Research Council. | ||||||
| * % at 4 years or beyond | ||||||
| EORTC/GIMEMA AML8 | 40 | 57 | 48 | 30 | 56 | 46 |
| GOELAM | NA | NA | 44 | 40 | 50 | 55 |
| MRC AML 10 | 37 | 58 P = 0.0007 | 53 | 40 P = 0.04 | 57 | 45 |
| Relapse* (%) | Disease-Free Survival* (%) | Overall Survival* (%) | ||||
|---|---|---|---|---|---|---|
| Trial | Autograft | Chemo | Autograft | Chemo | Autograft | Chemo |
| Abbreviations: EORTC/GIMEMA, European Organization for Research and Treatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; AML, acute myeloid leukemia; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; NA, not applicable; MRC, Medical Research Council. | ||||||
| * % at 4 years or beyond | ||||||
| EORTC/GIMEMA AML8 | 40 | 57 | 48 | 30 | 56 | 46 |
| GOELAM | NA | NA | 44 | 40 | 50 | 55 |
| MRC AML 10 | 37 | 58 P = 0.0007 | 53 | 40 P = 0.04 | 57 | 45 |
Cytogenetic classification for risk grouping in acute myeloid leukemia (AML).*†
| Cytogenetic Abnormality | Frequen cy in Children | Frequency in Adults | Fusion Genes | Genetic and Biologic Modifiers of Outcome |
|---|---|---|---|---|
| Others: 20q, 21q, del (9q), t(9;22), abn 17p, complex karyotypes (≥ 3) | ||||
| *Adapted from References 1, 2, 11, 24. | ||||
| †It should be noted that this classification scheme is that used by the US Cooperative Groups.24 The MRC11 uses an altered classification scheme in which abn11q23, del(9), del(7q) without other abnormalities and complex abn ≥ 3 but ≤ 5 are considered “intermediate” risk, not “unfavorable” as shown in this table. Awareness of these differences in cytogenetic risk grouping is critical for comparison of outcome on clinical trials conducted by these groups. | ||||
| “Favorable” Cytogenetics | ||||
| t(8;21)(q22;q22) | 12% | 5-8% (< 55 yrs), Rare (> 55 yrs) | AML1/ETO | FLT3 ITD (9%) + del(9q), + Complex |
| Inv(16)(p13q22) | 12% | 10% (< 45 yrs), Rare (> 45 yrs) | CBFβ/MYH11 | FLT3 ITD (7%) |
| t(16;16)(p13;q22) | ||||
| t(15;17)(q21;q11) | 7% | 15% (< 45 years), Rare (> 45 years) | PML-RARα | FLT3 ITD (37%) |
| Variants: | ||||
| t(11;17)(q23;q11) | PLZF-RARα | |||
| t(5;17)(q32;q11) | NPM-RARα | |||
| t(11;17)(q13;q11) | NuMA-RARα | |||
| “Intermediate” Cytogenetics | ||||
| +8 | Rare | 10% | FLT3 ITD (28%) | |
| Normal karyotype | 10-15% | 15-20% | FLT3 ITD (34%) | |
| MLL ITD (10%) | ||||
| Others: Y, +6, All other karyotypes not considered favorable or unfavorable | FLT3 ITD (20-30%) | |||
| “Unfavorable” Cytogenetics | ||||
| Abn 11q23 | > 50% of infant AML cases 7% t(9;11) | 5-7% | MLL | FLT3 ITD (0%) |
| Common Variants: | ||||
| t(4;11)(q21;q23) | MLL/AF4 | |||
| t(9;11)(p22;q23) | MLL/AF9 | |||
| t(11;19)(q23;p13.1) | MLL/ELL | |||
| t(11;19)(q23;p13.3) | MLL/ENL | |||
| t(6;9)(p23;q34) | Rare | <1% | DEK/CAN | Inv(3)(q21q26), |
| t(3;3)(q21;q26) | 3% | 3-5% | Ribophorin/EVI1 | FLT3 ITD (17%) |
| –5/del(5q) | Rare | <10% (< 45 yrs) >10% (> 45 yrs) | MDR drug resistance FLT3 ITD (0%) | |
| –7/del(7q) | 10% | <10% (< 45 yrs) >10% (> 45 yrs) | MDR drug resistance FLT3 ITD (7%) | |
| Cytogenetic Abnormality | Frequen cy in Children | Frequency in Adults | Fusion Genes | Genetic and Biologic Modifiers of Outcome |
|---|---|---|---|---|
| Others: 20q, 21q, del (9q), t(9;22), abn 17p, complex karyotypes (≥ 3) | ||||
| *Adapted from References 1, 2, 11, 24. | ||||
| †It should be noted that this classification scheme is that used by the US Cooperative Groups.24 The MRC11 uses an altered classification scheme in which abn11q23, del(9), del(7q) without other abnormalities and complex abn ≥ 3 but ≤ 5 are considered “intermediate” risk, not “unfavorable” as shown in this table. Awareness of these differences in cytogenetic risk grouping is critical for comparison of outcome on clinical trials conducted by these groups. | ||||
| “Favorable” Cytogenetics | ||||
| t(8;21)(q22;q22) | 12% | 5-8% (< 55 yrs), Rare (> 55 yrs) | AML1/ETO | FLT3 ITD (9%) + del(9q), + Complex |
| Inv(16)(p13q22) | 12% | 10% (< 45 yrs), Rare (> 45 yrs) | CBFβ/MYH11 | FLT3 ITD (7%) |
| t(16;16)(p13;q22) | ||||
| t(15;17)(q21;q11) | 7% | 15% (< 45 years), Rare (> 45 years) | PML-RARα | FLT3 ITD (37%) |
| Variants: | ||||
| t(11;17)(q23;q11) | PLZF-RARα | |||
| t(5;17)(q32;q11) | NPM-RARα | |||
| t(11;17)(q13;q11) | NuMA-RARα | |||
| “Intermediate” Cytogenetics | ||||
| +8 | Rare | 10% | FLT3 ITD (28%) | |
| Normal karyotype | 10-15% | 15-20% | FLT3 ITD (34%) | |
| MLL ITD (10%) | ||||
| Others: Y, +6, All other karyotypes not considered favorable or unfavorable | FLT3 ITD (20-30%) | |||
| “Unfavorable” Cytogenetics | ||||
| Abn 11q23 | > 50% of infant AML cases 7% t(9;11) | 5-7% | MLL | FLT3 ITD (0%) |
| Common Variants: | ||||
| t(4;11)(q21;q23) | MLL/AF4 | |||
| t(9;11)(p22;q23) | MLL/AF9 | |||
| t(11;19)(q23;p13.1) | MLL/ELL | |||
| t(11;19)(q23;p13.3) | MLL/ENL | |||
| t(6;9)(p23;q34) | Rare | <1% | DEK/CAN | Inv(3)(q21q26), |
| t(3;3)(q21;q26) | 3% | 3-5% | Ribophorin/EVI1 | FLT3 ITD (17%) |
| –5/del(5q) | Rare | <10% (< 45 yrs) >10% (> 45 yrs) | MDR drug resistance FLT3 ITD (0%) | |
| –7/del(7q) | 10% | <10% (< 45 yrs) >10% (> 45 yrs) | MDR drug resistance FLT3 ITD (7%) | |
Genes shared and distinctive between MLL translocation variants (MLL/ALL1/HRX gene on chromosome 11q23) in acute myeloid leukemia (AML)/acute lymphoblastic leukemia (ALL).
| Genes Shared in All MLL Variants | t(4;11) | t(11;19) | t(9;11) | t(10;11) |
|---|---|---|---|---|
| † FLT3 expression is more significantly associated with t(4;11) than with all MLL variants. | ||||
| Lyn | Bmi-1 | HRAFY | TRADD | RUNX3 |
| NFATC3 | DOK1 | ERH | TCFL4 | HMGCR |
| RAD9 | WAS | IL2Rγ | COX7 | HGF |
| CD44 | PPKAR | IRAK1 | HSF1 | CDKN1CD |
| AC133 | YES | IGBP1 | AHR | RARα |
| BAG1 | TIMP1 | TACT | PLCγ2 | RARES2 |
| CRADD | ELF4 | LPXN | Rab33A | TNFSF9 |
| Blk | JUND | Serpine | RASA1 | GIT2 |
| FLT3† | FLT3† | MAGED2 | DOC1R | SH3BP1 |
| Genes Shared in All MLL Variants | t(4;11) | t(11;19) | t(9;11) | t(10;11) |
|---|---|---|---|---|
| † FLT3 expression is more significantly associated with t(4;11) than with all MLL variants. | ||||
| Lyn | Bmi-1 | HRAFY | TRADD | RUNX3 |
| NFATC3 | DOK1 | ERH | TCFL4 | HMGCR |
| RAD9 | WAS | IL2Rγ | COX7 | HGF |
| CD44 | PPKAR | IRAK1 | HSF1 | CDKN1CD |
| AC133 | YES | IGBP1 | AHR | RARα |
| BAG1 | TIMP1 | TACT | PLCγ2 | RARES2 |
| CRADD | ELF4 | LPXN | Rab33A | TNFSF9 |
| Blk | JUND | Serpine | RASA1 | GIT2 |
| FLT3† | FLT3† | MAGED2 | DOC1R | SH3BP1 |
Genes and pathways associated with treatment failure in infant leukemia
| Genes and Pathways | Individual Genes |
|---|---|
| DNA repair pathways | Helicase RecQ1 RAD1 |
| Chromatin methylation | Methyltransferases |
| Signal transduction | TGFβ signaling pathways |
| Ras signaling pathways | Farnesyl transferase |
| PI3 Kinase | |
| PLCε | |
| RANTES | |
| RAS | |
| DYRK3 | |
| TGFβ signaling pathways | BMP 1, 2, 4 |
| TGFβ binding proteins | |
| Interferon-stimulated genes |
| Genes and Pathways | Individual Genes |
|---|---|
| DNA repair pathways | Helicase RecQ1 RAD1 |
| Chromatin methylation | Methyltransferases |
| Signal transduction | TGFβ signaling pathways |
| Ras signaling pathways | Farnesyl transferase |
| PI3 Kinase | |
| PLCε | |
| RANTES | |
| RAS | |
| DYRK3 | |
| TGFβ signaling pathways | BMP 1, 2, 4 |
| TGFβ binding proteins | |
| Interferon-stimulated genes |
Prognosis in acute myeloid leukemia (AML) by leukemia karyotype.
| Karyotype | % | % CR | % EFS |
|---|---|---|---|
| Abbreviations: EFS, event-free survival. | |||
| Favorable | |||
| t(8;21) | 5-10 | 90 | 50-70 |
| inversion | 16 | 5-10 | 90 50-70 |
| t(15;17) | 5-10 | 80-90 | 70 |
| Intermediate | |||
| diploid, -Y | 40-50 | 70-80 | 20-40 |
| Unfavorable | |||
| -5/-7 | 20-30 | 40 | 5-10 |
| +8 | 10 | 60 | 10-20 |
| 11q23,20q-, other | 10-120 | 60 | 10 |
| Karyotype | % | % CR | % EFS |
|---|---|---|---|
| Abbreviations: EFS, event-free survival. | |||
| Favorable | |||
| t(8;21) | 5-10 | 90 | 50-70 |
| inversion | 16 | 5-10 | 90 50-70 |
| t(15;17) | 5-10 | 80-90 | 70 |
| Intermediate | |||
| diploid, -Y | 40-50 | 70-80 | 20-40 |
| Unfavorable | |||
| -5/-7 | 20-30 | 40 | 5-10 |
| +8 | 10 | 60 | 10-20 |
| 11q23,20q-, other | 10-120 | 60 | 10 |
Outcome in acute myeloid leukemia (AML) with t(8;21) by number of high-dose ara-C consolidations.*
| Number of High-Dose Ara-C Courses | |||
|---|---|---|---|
| Parameter | 1 | ≥ 3 | Pvalue |
| *Data from Byrd et al.26 | |||
| Abbreviations: CR, complete remission; DFS, disease-free survival. | |||
| Relapse, % | 62 | 19 | 0.004 |
| Median CR duration (mos) | 10.5 | > 35 | |
| 5-year DFS, % | 38 | 61 | 0.03 |
| Median survival (mos) | 24 | > 43 | |
| 5-year survival, % | 44 | 76 | 0.04 |
| Number of High-Dose Ara-C Courses | |||
|---|---|---|---|
| Parameter | 1 | ≥ 3 | Pvalue |
| *Data from Byrd et al.26 | |||
| Abbreviations: CR, complete remission; DFS, disease-free survival. | |||
| Relapse, % | 62 | 19 | 0.004 |
| Median CR duration (mos) | 10.5 | > 35 | |
| 5-year DFS, % | 38 | 61 | 0.03 |
| Median survival (mos) | 24 | > 43 | |
| 5-year survival, % | 44 | 76 | 0.04 |
Topotecan + ara-C and fludarabine + ara-C regimens in acute myeloid leukemia (AML).
| A. Regimen Schedule | |||
|---|---|---|---|
| 1. | IA ara-C | IDA 1.5 g/m2/CI/D × 3-4 | 12 mg/m2/D × 3 |
| 2. | FAIA ara-C | fludarabine 2 g/m2 over 4 h/D × 5 | 30 mg/m2/D × 5 |
| 3. | TA ara-C | Topotecan 1-2 g/m2 over 2-4 h/D × 5 | 1.25 mg/m2 CI/D × 5 |
| A. Regimen Schedule | |||
|---|---|---|---|
| 1. | IA ara-C | IDA 1.5 g/m2/CI/D × 3-4 | 12 mg/m2/D × 3 |
| 2. | FAIA ara-C | fludarabine 2 g/m2 over 4 h/D × 5 | 30 mg/m2/D × 5 |
| 3. | TA ara-C | Topotecan 1-2 g/m2 over 2-4 h/D × 5 | 1.25 mg/m2 CI/D × 5 |
| B. Results | ||||
|---|---|---|---|---|
| Regimen | No. | % CR | EFS | Median Survival (wks) |
| Abbreviations: IDA indicates idarubicin; CI, confidence interval; CR, complete remission; EFS, event-free survival. | ||||
| IA | 322 | 77 | 63 | 77 |
| FA | 600 | 55 | 40 | 30 |
| TA | 357 | 59 | 36 | 41 |
| B. Results | ||||
|---|---|---|---|---|
| Regimen | No. | % CR | EFS | Median Survival (wks) |
| Abbreviations: IDA indicates idarubicin; CI, confidence interval; CR, complete remission; EFS, event-free survival. | ||||
| IA | 322 | 77 | 63 | 77 |
| FA | 600 | 55 | 40 | 30 |
| TA | 357 | 59 | 36 | 41 |
Preliminary results of the Phase II study of clofarabine in refractory relapse leukemia.
| Disease | No. Evaluable | No. CR + CRp (%) |
|---|---|---|
| Abbreviations: AML, acute myeloid leukemia; ALL, acute lymphocytic leukemia; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; CR, complete remission; CRp, CR with low platelets. | ||
| AML | 20 | 11 + 1 (60) |
| ALL | 6 | 1 + 1 (33) |
| CML-blastic phase | 6 | 3 + 1 (67) |
| MDS/CML | 4 | 1 + 2 (75) |
| Disease | No. Evaluable | No. CR + CRp (%) |
|---|---|---|
| Abbreviations: AML, acute myeloid leukemia; ALL, acute lymphocytic leukemia; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; CR, complete remission; CRp, CR with low platelets. | ||
| AML | 20 | 11 + 1 (60) |
| ALL | 6 | 1 + 1 (33) |
| CML-blastic phase | 6 | 3 + 1 (67) |
| MDS/CML | 4 | 1 + 2 (75) |
Results of GO ± IL-11 therapy in newly diagnosed acute myeloid leukemia (AML)/high-risk myelodysplastic syndrome (MDS) patients at high risk of mortality from chemotherapy regimen.76
| No. Treated | No. CR (%) | No. Induction Death (%) | |
|---|---|---|---|
| *Data from Estey et al.39 | |||
| Abbreviations: IL-11, interleukin-11; CR, complete remission; GO, gemtuzumab ozogamicin. | |||
| Total | 54 | 8 (15) | 20 (37) |
| Karyotype | |||
| diploid | 31 | 6 (19) | 6 (19) |
| other | 23 | 2 (9) | 14 (61) |
| Therapy | |||
| GO | 26 | 0 (0) | 12 (46) |
| GO + IL-11 | 28 | 8 (29) | 8 (29) |
| No. Treated | No. CR (%) | No. Induction Death (%) | |
|---|---|---|---|
| *Data from Estey et al.39 | |||
| Abbreviations: IL-11, interleukin-11; CR, complete remission; GO, gemtuzumab ozogamicin. | |||
| Total | 54 | 8 (15) | 20 (37) |
| Karyotype | |||
| diploid | 31 | 6 (19) | 6 (19) |
| other | 23 | 2 (9) | 14 (61) |
| Therapy | |||
| GO | 26 | 0 (0) | 12 (46) |
| GO + IL-11 | 28 | 8 (29) | 8 (29) |
Response and toxicity results of daunorubicin + ara-C ± cyclosporin A in acute myeloid leukemia (AML).*
| Parameter | Number | Yes |
|---|---|---|
| *Data from List et al.58 | ||
| Complete remission, % | 33 | 39 |
| Death, % | 18 | 14 |
| ↑ Bili grade 4, % | 8 | 30 |
| 2-year relapse-free survival, % | 9 | 34 |
| 2-year survival, % | 12 | 22 |
| Parameter | Number | Yes |
|---|---|---|
| *Data from List et al.58 | ||
| Complete remission, % | 33 | 39 |
| Death, % | 18 | 14 |
| ↑ Bili grade 4, % | 8 | 30 |
| 2-year relapse-free survival, % | 9 | 34 |
| 2-year survival, % | 12 | 22 |
Results of azacytidine (AZA) versus supportive care in myelodysplastic syndrome (MDS).*
| AZA | Supportive Care | PValue | |
|---|---|---|---|
| *Data from Silverman et al.92 | |||
| Number treated | 99 | 92 | |
| Complete remission, % | 6 | 0 | |
| Partial remission/improved, % | 10/47 | 0/7 | <0.001 |
| Median time to transformation (mos) | 22 | 12 | 0.003 |
| Median survival (mos) | 18 | 14 | 0.1 |
| AZA | Supportive Care | PValue | |
|---|---|---|---|
| *Data from Silverman et al.92 | |||
| Number treated | 99 | 92 | |
| Complete remission, % | 6 | 0 | |
| Partial remission/improved, % | 10/47 | 0/7 | <0.001 |
| Median time to transformation (mos) | 22 | 12 | 0.003 |
| Median survival (mos) | 18 | 14 | 0.1 |

Interactions of normal and leukemic progenitors with the hematopoietic microenvironment (HM).
Abbreviations: SDF, stromal-derived factor; VLA, very late antigen; LFA, lymphocyte function antigen; CAM, cell adhesion molecule; VCAM, vascular adhesion molecule; SCF, stem cell factor.
Interactions of normal and leukemic progenitors with the hematopoietic microenvironment (HM).
Abbreviations: SDF, stromal-derived factor; VLA, very late antigen; LFA, lymphocyte function antigen; CAM, cell adhesion molecule; VCAM, vascular adhesion molecule; SCF, stem cell factor.

MRC AML 12: Overall survival on a donor versus no donor basis.
Figure presented with permission of Prof. Burnett, chairman of the MRC AML trials.
MRC AML 12: Overall survival on a donor versus no donor basis.
Figure presented with permission of Prof. Burnett, chairman of the MRC AML trials.

MRC AML 10: Disease-free survival in patients treated with autologous bone marrow transplantation (ABMT) versus no further treatment.
Abbreviations: CR, complete remission.
Reprinted with permission of Prof. AK Burnett and The Lancet. 1998 Mar 7; 351(9104):700-8.
MRC AML 10: Disease-free survival in patients treated with autologous bone marrow transplantation (ABMT) versus no further treatment.
Abbreviations: CR, complete remission.
Reprinted with permission of Prof. AK Burnett and The Lancet. 1998 Mar 7; 351(9104):700-8.

Eastern Cooperative Oncology Group (ECOG) protocol trial overview.
Abbreviations: Ara-C, cytarabine; CR, complete remission; HDAC, high-dose Ara-C; PBSC, peripheral blood stem cell.
Eastern Cooperative Oncology Group (ECOG) protocol trial overview.
Abbreviations: Ara-C, cytarabine; CR, complete remission; HDAC, high-dose Ara-C; PBSC, peripheral blood stem cell.

AML 15 protocol flowchart.
Abbreviations: AML, acute myeloid leukemia; DAT, daunorubicin, cytarabine, thioguanine; CR, complete remission; Mab, monoclonal antibody; FLAG, fludarabine, cytarabine; Ida, Idarubicin; allo SCT, allogenic stem cell transplantation; ARA-C, cytarabine.
AML 15 protocol flowchart.
Abbreviations: AML, acute myeloid leukemia; DAT, daunorubicin, cytarabine, thioguanine; CR, complete remission; Mab, monoclonal antibody; FLAG, fludarabine, cytarabine; Ida, Idarubicin; allo SCT, allogenic stem cell transplantation; ARA-C, cytarabine.

