Abstract
Severe congenital neutropenias (SCNs) are rare diseases, and to date about 30 subtypes have been described according to their genetic causes. Standard care aims to prevent infections and limit the risk of leukemic transformation; however, several subtypes may have additional organ dysfunction(s), requiring specialized care. Granulocyte colony-stimulating factor and hematopoietic stem cell transplantation are now the bedrock of standard care. Better understanding of SCN mechanisms now offers the possibility of adapted therapy for some entities. An inhibitor of sodium glucose cotransporter, an antidiabetic drug, may attenuate glycogen storage disease type Ib and glucose-6-phosphatase catalytic subunit 3 neutropenias by clearing 1,5-anhydroglucitol, the precursor of the phosphate ester responsible for these SCNs. Chemokine receptor CXCR4 inhibitors contribute to reversing the leukocyte defect in warts, hypoglobulinemia, infections, and myelokathexis syndrome. All these new approaches use oral drugs, which notably improve quality of life. Additionally, improved research into clonal evolution has highlighted some ways to potentially prevent leukemia, such as stimulating somatic genetic rescue, a physiological process that might limit the risk of leukemic transformation.
Learning Objectives
Identify in the clinical presentation of a patient with neutropenia the key features suggesting a possible genetic cause
Realize that in G6PC3 neutropenia and in GSDIB, gliflozine may be considered an alternative to standard care with GCSF and/or HSCT
Learn that in CXCR4 WHIM syndrome, CXCR4 inhibitors offer a therapeutic approach
Introduction
Interest in severe congenital neutropenias (SCNs) was heightened by the availability of a new therapeutic agent, granulocyte colony-stimulating factor (GCSF), at the beginning of the 1990s.1 It is an understatement to say that GCSF dramatically transformed hematology medical practice. Even though GCSF is mainly indicated for chemotherapy- induced neutropenia, it has also focused light on the extremely rare group of diseases called SCNs. GCSF marketing was associated with a recommendation by health authorities to develop patient registries, primarily to assess its potential leukemic risk. Incidentally, registry studies have provided valuable evidence not only to demonstrate the efficacy of GCSF but also to show that high doses induce clonal hematopoiesis, leading to myelodysplasia or acute leukemia.2,3 We now know that this risk can be limited by hematopoietic stem cell transplantation (HSCT) early during SCN evolution.4 But even if HSCT can control the risk of myelodysplastic syndrome and even if GCSF remains effective throughout a patient's lifetime, daily GCSF use engenders quality-of-life limitations, explaining poor compliance and thus, only partial efficacy, with sequelae, persistent symptoms, and sometimes life-threatening infections.5 Such limitations are strong motivations to find new therapeutic approaches. And genetic analyses have opened the gate to potential novel agents.
SCN genetics: accurate and quick diagnoses for better classification and more therapeutic options
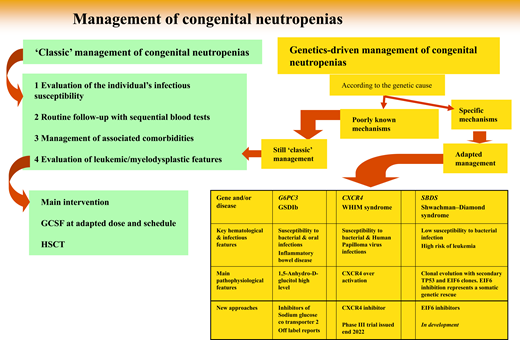
Since 1993, while registries were being progressively created, genetic technologies have advanced dramatically, and about 30 distinct SCN genetic entities have been described (Table 1).6 Above all, genetics provides a reliable parameter to classify a patient's disease. Initially, determining a culprit gene derived from the laborious collection of biological material from phenotypically well-defined patients. Elastase, neutrophil expressed (ELANE) was the first gene identified,7 followed by the Shwachman-Bodian-Diamond syndrome (SBDS) gene.8 But once the medical community had access to more powerful genetic tools, such as targeted next generation sequencing and whole-exome sequencing, it became possible to study simultaneously the same panel of genes involved in this group of diseases regardless of the patient's phenotype. This powerful approach broke the lines separating classic phenotypes.9 Some entities may share symptoms attributable to different genes, while very different clinical manifestations can be caused by the same genetic mutation(s). As a consequence, the term “SCN” is not completely accurate, and perhaps calling these entities genetic neutropenias would be more appropriate. Notably, genetic neutropenias (GNs) are not always severe, and the adjective “severe” is not justified by many patients' clinical status. Now, obviously, genetic expertise is the key to diagnosis. Although blood tests have become routine examinations, physicians must justify genetic testing, as it is widely accepted for only a restricted number of settings.
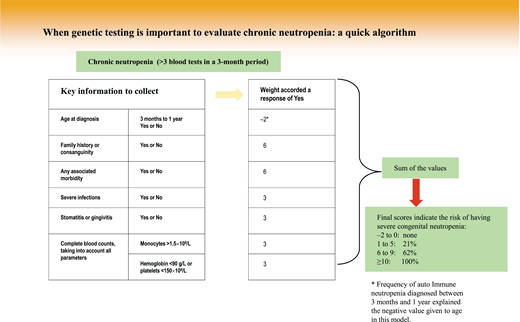
Indeed, population study results have shown that neutropenia is very common. Depending on geographic origin, neutropenia is detected in 1% (Caucasians) to 10% (those with African origins) of people, while GN prevalence is less than 1 in 10 000.10,11 This situation explains why a clinical algorithm makes it possible to restrict genetic testing to suspected cases in an attempt to limit patient anxiety and avoid waste of medical resources. One algorithm, very easy to use, was reported for pediatric neutropenia, and its score can even be computed during a short phone call to specify the patient's characteristics. A rapid decision of whether or not to launch genetic research can then be made by calculating the score of the individual algorithm elements (Figure 1).12
From genetic diagnosis to therapeutic approaches: 3 situations
But once a genetic diagnosis is fully determined, how can this information change the natural history of a patient's disease? Here we develop 3 situations (4 diseases) in which genetic information, based on a large amount of basic scientific research, deciphers the molecular consequences of a genetic defect and leads to a specific, personalized therapeutic approach not yet at the same stage of development.
Inhibitor of sodium glucose cotransporter to treat G6PC3 and GSDIB SCNs
Glucose-6-phosphatase catalytic subunit 3 (G6PC3) and glycogen storage disease type Ib (GSDIB) neutropenias are caused by a defect in the glucose-6-phosphatase (G6Pase) enzyme.13,14 For years, the mechanism of neutropenia in those entities remained unclear. The main enigma was the lack of a direct role of G6Pase, which is composed of several units (ie, the endoplasmic reticulum (ER)/cytosol transporter in GSDIB and the catalytic subunit 3 in G6PC3 GNs) in the neutrophils' metabolic machinery. Even if a genetic defect was clearly documented, a key was missing to understand these 2 diseases because G6Pase did not appear to be responsible for a direct defect in the neutrophil-energy pathway. This mystery was solved when the role of 1,5-anhydroglucitol phosphate was identified, and the 2 defect proteins involved in GSDIB and in G6PC3 GN were shown to play an important role in the removal of a toxic metabolite, 1,5-anhydroglucitol-6- phosphate (1,5AG6P), in neutrophils.15 1,5AG6P is produced by phosphorylation of 1,5-anhydroglucitol (1,5AG), a glucose analogue derived from food and normally present in the plasma. As 1,5AG6P is an inhibitor of hexokinase, the enzyme that allows glucose metabolism by glycolysis, it has to be destroyed as soon as it is formed. This is the function of G6PC3, an enzyme related to the classical G6Pase but that has only little G6Pase activity. The transport of 1,5AG6P from the cytosol to the lumen of the ER is carried out by the glucose-6-phosphate transporter encoded by SLC37A4, the gene mutated in GSDIB. The cytosolic accumulation of 1,5AG6P due to mutations in G6PC3 or in SLC37A4 leads to the inhibition of glycolysis. This mechanism explains neutrophil dysfunction and apoptosis in GSDIB and G6PC3-deficiency GNs.15 That finding had potential clinical impact, as it opened the way to reverse the clinical manifestations of neutropenia and neutrophil dysfunction in GSDIB and G6PC3-deficient patients. Indeed, prescribed off- label, inhibitors of sodium glucose cotransporter (eg, empagliflozin or dapagliflozin) that are routinely used clinically as antidiabetic drugs to treat type 2 diabetes inhibit renal glucose uptake, cause glycosuria, block renal 1,5AG reabsorption, and lower plasma glucose (to the renal glucose threshold) and 1,5AG concentrations. Empagliflozin was also shown to decrease intracellular 1,5AG6P in G6PC3-deficient mice and normalize their absolute neutrophil counts.15 That finding has now been confirmed in humans16 : the clinical use of inhibitors of sodium-glucose linked transporter type 2 did not interfere with patients' glucose levels (which, in this context, are low or normal) but increased 1,5AG clearance and allowed myeloid maturation and function. For the first 4 reported cases,16 all with GSDIB, empagliflozin, during short-term follow-up of less than 1 year, demonstrated both diminished 1,5AG and that GCSF support could be dramatically lowered or even withdrawn while infectious events were controlled.17 Additional cases with longer follow-up have confirmed those observations and offer a cheaper approach and much more efficient management of both GSDIB and G6PC3 GNs.18,19
CXCR4 inhibitors and WHIM syndrome
Warts, hypoglobulinemia, infections, and myelokathexis (WHIM) syndrome is a very rare GN caused by a mutation of the CXCR4 chemokine receptor. In 1964, Zuelzer and Krill described an exceptional congenital neutropenia associated with bone marrow (BM) hyperplasia of mature neutrophils: myelokathexis.20 In 1990, Wetzler proposed the acronym WHIM, reflecting the manifestations of human papillomavirus (HPV)-induced warts, hypogammaglobulinemia, and bacterial infections together with myelokathexis. WHIM syndrome is characterized by heterogeneous disease manifestations that include severe infectious episodes, HPV-associated warts, panleukopenia, and hypogammaglobulinemia.21,22 Its clinical onset and complications are more variable than originally suspected. Neutropenia is associated with lymphopenia and monocytopenia, which are almost always observed in patients suffering from this disorder; HPV infections are extremely common. WHIM syndrome is a potentially fatal disease mostly because of immune-system alterations and HPV infections.21,22
Genetic analyses of WHIM syndrome patients identified inherited heterozygous autosomal dominant mutations in the CXCR4 gene encoding the receptor of the CXC α-chemokine (CXCL12),23 which regulates hematopoiesis and the peripheral trafficking of neutrophil and lymphocyte subsets. CXCR4 was first studied for its role as a human immunodeficiency virus coreceptor. The functional consequences of the variants are dramatic, as they confer a CXCR4 gain of function responsible for the WHIM syndrome–associated panleukopenia. Current WHIM syndrome therapies are intravenous immunoglobulin and GCSF injections and antibiotic prophylaxis.24 Those treatments may limit the severe infections affecting patients, but they fail to control potentially lethal HPV or mycobacterial infections. Since the genetic findings showed CXCR4 gain of function, agents inhibiting CXCR4 function appear promising. Those treatments, initially developed to control HIV infection, were obviously abandoned but were later shown to possibly act as a stem cell mobilizer.
Indeed, the AMD3100 compound was patented under the name Plerixafor to mobilize autograft stem cells. Successfully tested on a very limited number of WHIM syndrome patients, that drug efficiently mobilized granulocytes, lymphocytes, and monocytes,25,26 and even controlled HPV infections, in 3 patients.27 Plerixafor development was not pursued, but another compound, mavorixafor, was developed by another company. After phase 1 and phase 2 studies,28 that latter molecule is now being evaluated in a phase 3 double-blind randomized trial; its results are expected by the end of 2022. The findings of the phase 2 study on mavorixafor showed that neutrophil and, in general, all leukocyte defects were corrected by this oral drug, with the added advantage of limiting HPV infections.28 Notably, because CXCR4/CXCL12 interacts strongly with the GCSF-3 receptor (CSF3R) axis early during myelopoiesis, inhibiting CXCR4 enables neutrophil mobilization via a pathway other than GCSF. And now an oral compound may become available to avoid neutropenia in other chronic subtypes as well as post chemotherapy.
Somatic genetic rescue: clonal hematopoiesis is not always bad news
In addition to a high risk of infections, almost all GNs carry an elevated risk of leukemic transformation, for which the percentage depends on the gene involved. The trajectory from the naive germ line at birth to leukemia is a multistep mutational process. The high frequency of clonal evolution can be viewed as premature aging of clonal hematopoiesis, but clonal architecture is strongly dependent on the germline variant on which it develops: clonal hematopoiesis differs markedly in ELANE neutropenia, Shwachman-Diamond syndrome (SDS), and GATA2 syndrome.29-31
SDS, a recessive multisystem disorder characterized by exocrine pancreas deficiency, mild neutropenia, and various other organ dysfunctions, is caused by compound heterozygous mutations of the SBDS gene and offers an initial insight into the possible role of somatic genetic rescue to prevent leukemic transformation.32 The SBDS protein is an essential cofactor for elongation factor-1. Biallelic SBDS mutations impair the release of antiassociation, eukaryotic translation-initiation factor-6 (EIF6) from the 60S ribosomal unit. The cell tries to escape that blockage in different ways. One quite deleterious method that directly involves the leukemic outcome is the overstimulation of the TP53 pathway,33 induced by ribosomal stress.34 The involvement of TP53 mutations in SDS has been documented several times during SDS patients' lifetimes. During the “chronic phase,” the TP53 clone frequently has a low variant-allele frequency, and at myelodysplastic syndrome onset, the TP53 variant-allele frequency is high, typically with biallelic variants.30,35 But it is not the sole mutational event observed during the course of SDS. The second frequent molecular event concerns the EIF6 variant.30 The occurrence of such a molecular event was suggested by a common cytogenetic finding in SDS patients' BM: the del20q clone, which harbors EIF6. But other molecular events (eg, point mutations, reciprocal chromosomal translocation) limit EIF6 concentration or EIF6 binding to the 60S subunit and reverse the ribosome-assembly and protein-synthesis defects.36 Such mechanisms suggest that EIF6 inhibition might constitute a valuable strategy to compensate for the SBDS defect and represents a promising therapeutic strategy for SDS.36,37
CLINICAL CASE
The patient is an Algerian-born male, issued from a consanguineous family, who has previously been reported in a case series (as patient 5643).38 He was first seen in our unit at the age of 7 years, when he had already experienced about 12 distinct severe infectious episodes, including bacterial meningitis and 3 types of pneumonitis since birth in addition to recurrent oral infections. He failed to thrive and had chronic diarrhea. Morphologically, he had a narrow thorax and thin skin with a prominent superficial venous network on the limbs and the abdomen. Echocardiography detected aortic insufficiency, without hemodynamic consequences. Initial blood tests showed profound neutropenia (absolute neutrophil count, 240/mm3 ), with mild anemia (hemoglobin, 9.8 g/dL) and a normal platelet count. His first BM cytology did not reveal any significant abnormalities, with a myeloid to nucleated erythroid cell ratio of 5:1 and no maturation arrest. At that time, the sole diagnostic genetic test available was for ELANE, and no such variants were identified. His chronic diarrhea was difficult to analyze because of the presence of steatorrhea and a fat-soluble-vitamin deficiency but showed no documented exocrine pancreas deficiency. GCSF (5-7 µg/kg/d) was prescribed, along with pancreatic enzyme and nutritional support. The patient returned to Algeria and continued to be treated between there and France. Repeated blood counts showed recurrent thrombocytopenia. Endoscopy of the digestive tract at 10 years old found inflammation; inflammatory bowel disease was diagnosed and treated with steroids. This situation, with recurrent infections and difficulty obtaining GCSF, resulted in several infections, including a colonic abscess and a colonic fistula necessitating a hemicolectomy that incurred major surgical complications. Although a molecular diagnosis of SBDS mutation was sought when he was 10 years old, it excluded a pathogenic variant; G6CP3-deficient neutropenia was finally diagnosed when he was 15 years old as the disease had just been identified by Sanger sequencing.13
After developing a gut fistula at 18 years of age, he returned to France. A prescription of anti–tumor necrosis factor α (TNF-α) for about 5 years, combined with long-term GCSF, maintained a limited quality of life, as his disease affected his ability to go to school and later work. At the age of 23 years, he was prescribed dapagliflozin at the initial dose of 0.2 mg/kg in addition to his long-term therapy. Figure 2 shows his neutrophil, platelet, and blood 1,5AG evolutions over time. He initially received GCSF (9 µg/kg) 3 times per week in conjunction with anti-TNF-α. Because of a partial neutrophil count and 1.5AG-clearance responses, the dapagliflozin dose was progressively increased up to 0.9 mg/kg/d. Oral dapagliflozin has been continued since then (he is now 25 years old) for a total of 16 months. Notably, after 6 months the anti-TNF-α was withdrawn, and the inflammatory bowel disease did not recur. An attempt to stop GCSF was complicated by an oral infection; however, it has now been tapered down to 3 µg/kg twice weekly.

A visual summary of the medical history of a patient with G6PC3 neutropenia treated by gliflozine.
A visual summary of the medical history of a patient with G6PC3 neutropenia treated by gliflozine.
Conclusion
The standard care of GNs is based on GCSF and HSCT. Driven by the extensive development of genetic background analyses of such entities, better understanding of the mechanisms at work now offers some possibilities of adapted therapy. ISGTL2, an antidiabetic drug, may partially reverse GSDIB and G6PC3 GNs by clearing 1,5AG, which is responsible for the associated neutropenia. CXCR4 inhibitors contribute to reversing the leukocyte defect in WHIM syndrome. Concerning the leukemic transformation risk, better understanding of the clonal evolution raises the possibility of preventing leukemia by stimulating somatic genetic rescue, a physiological process that might limit the risk of such progression.
Conflict-of-interest disclosure
Jean Donadieu: no competing financial interests to declare.
Christine Bellanné-Chantelot: no competing financial interests to declare.
Off-label drug use
Jean Donadieu: off-label drugs discussed are empagliflozin (Jardiance) and dapagliflozin (Forxiga).
Christine Bellanné-Chantelot: off-label drugs discussed are empagliflozin (Jardiance) and dapagliflozin (Forxiga).