Abstract
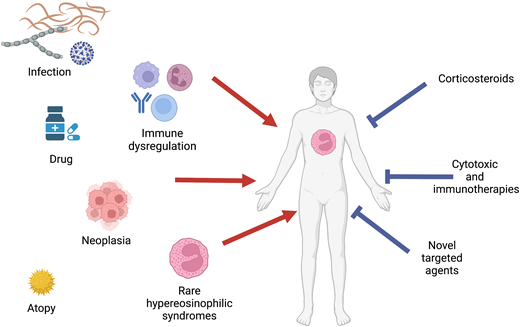
Hypereosinophilic syndromes (HES) are a heterogenous group of rare disorders with clinical manifestations ranging from fatigue to life-threatening endomyocardial fibrosis and thromboembolic events. Given the broad differential diagnosis of HES, a comprehensive approach is needed to identify potential secondary (treatable) causes and define end-organ manifestations. Classification by clinical HES subtype is also useful in terms of assessing prognosis and guiding therapy. Corticosteroids remain the mainstay of initial therapy in the setting of acute, life-threatening PDGFR mutation-negative HES. Whereas the recent availability of eosinophil-targeted therapies with extraordinary efficacy and little apparent toxicity is changing the treatment paradigm, especially for idiopathic HES and overlap syndromes, questions remain unanswered regarding the choice of agent, impact of combination therapies, and long-term effects of eosinophil depletion. This review provides a case-based discussion of the differential diagnosis of HES, including the classification by clinical HES subtype. Treatment options are reviewed, including novel eosinophil-targeted agents recently approved for the treatment of HES and/or other eosinophil-associated disorders. Primary (myeloid) disorders associated with hypereosinophilia are not be addressed in depth in this review.
Learning Objectives
To describe the heterogeneity of clinical presentations of hypereosinophilia
To discuss the approach to targeted therapy of hypereosinophilic syndrome
CLINICAL CASE
A 38-year-old previously healthy woman presented with intermittent but intense pruritus without rash. The pruritus initially involved only her ankles but subsequently spread up her legs with accompanying angioedema of the thighs and buttocks that prevented her from being able to wear pants. Antihistamines were ineffective, and a short course of solumedrol prescribed for sinusitis did not relieve the pruritus or swelling. A dermatologist prescribed oral and topical antibiotics for presumed folliculitis without improvement. One year after the onset of the symptoms, a complete blood count was performed and revealed anemia, thrombocytopenia (platelets 34 000), and eosinophilia (14.0 × 109/L). She was referred to hematology.
Differential diagnosis and initial evaluation
Eosinophilia, defined as an absolute eosinophil count (AEC) >0.45 × 109/L, is quite common, occurring in 1% to 2% of the general population.1 In contrast, hypereosinophilia (HE; AEC ≥1.5 × 109/L) is extremely rare, with an estimated incidence of 0.315 to 6.3 per 100 000 in the United States.2 The potential etiologies of eosinophilia (including HE) are varied and include allergic, infectious, neoplastic, genetic, and immune disorders (Table 1). Moreover, clinical symptoms are extremely heterogeneous. Dermatologic, pulmonary, and gastrointestinal manifestations are most frequently reported, but any organ system can be affected, and progression can occur over time without effective therapy.3 A careful and complete history and physical examination, including prior complete blood counts (if available), medication and travel history, assessment of cancer risk factors, and family history, is essential to narrow the differential. Initial laboratory and diagnostic testing should include complete blood count with differential, routine chemistries, serum immunoglobulin levels, B12 and tryptase, and assessment of lymphocyte clonality and phenotype. If there is a possible history of Strongyloides exposure, no matter how remote, serologic testing should be performed and/or empiric ivermectin (150 µg/kg × 1 dose) administered to prevent potentially fatal hyperinfection syndrome. Bone marrow biopsy and chest/abdomen/pelvis imaging should be strongly considered in any patient with AEC ≥1.5 × 109/L and no clear secondary cause of the HE. Additional testing, including testing for parasitic infections other than Strongyloides, should be guided by the clinical history and disease manifestations.
CLINICAL CASE (Continued)
A bone marrow biopsy specimen was hypercellular with increased eosinophils and plasma cells. Testing for FIP1L1::PDGFRA was negative. She was treated with prednisone 60 mg daily for presumed idiopathic thrombocytopenic purpura with clinical and hematologic improvement. However, as the prednisone was tapered, the eosinophilia, pruritus, and edema returned. She was referred to Mayo Clinic for further evaluation, which included an indeterminate serologic test for Strongyloides. She was treated with 2 doses of ivermectin and a 2-week course of albendazole. During this time, the prednisone was slowly tapered despite a rising AEC, peaking at 26.0 × 109/L on prednisone 5 mg every other day, and recurrent symptoms. The prednisone dose was increased to 25 mg daily, and hydroxyurea therapy (500 mg twice daily) was initiated. This was ineffective, and she was referred to the National Institutes of Health for further evaluation.
At the time of referral, she complained of fatigue, swelling, and extreme pruritus. Physical examination revealed symmetric nonpitting edema of the thighs and excoriations predominantly on the lower legs. Laboratory testing was notable for AEC 3.01 × 109/L; platelets 114 000; markedly elevated IgG, IgM, and IgE; and normal serum B12 and tryptase. Computed tomography (CT) scan was notable for borderline splenomegaly and minimal diffuse lymphadenopathy. Repeat bone marrow again showed only increased eosinophils. Mast cells were not increased, and testing for D816V KIT was negative. T-cell receptor testing by polymerase chain reaction showed a clonal pattern, and flow cytometry was notable for an aberrant CD3–CD4+CD10+ T-cell population, consistent with a diagnosis of lymphocytic variant hypereosinophilic syndrome (HES). B-cell clonality studies were negative. She was started on interferon α 1 mU daily with resolution of eosinophilia, platelets >50 k and symptomatic improvement.
Definition and classification of HES
The definition of HES has evolved over time since Chusid's landmark description of 14 patients with idiopathic HE and varied clinical manifestations in 1975.4 Whereas the current World Health Organization definition uses the term HES to describe only idiopathic HE with clinical manifestations,5 a recently updated consensus definition provides a broader approach that recognizes the overlap in clinical presentation between idiopathic and other types of HES, the imperfect sensitivity and specificity of available diagnostic testing, and the identification of new etiologies of HES over time6 (Table 2). In this consensus definition, a diagnosis of HE requires AEC >1.5 × 109/L on 2 examinations at least 1 month apart (to exclude laboratory error but allow diagnosis without a delay of 6 months) and/or tissue HE (to recognize the arbitrary nature of the AEC cutoff in the setting of clear eosinophil-mediated disease). HES is defined as HE with evidence of end-organ dysfunction attributable to the eosinophilia, irrespective of the cause. To address the heterogeneity of disorders included in this umbrella definition of HES and help guide diagnostic procedures and therapeutic choices, the following clinical subtypes have been proposed (Table 3)7 : (1) myeloid HE/HES (suspected or proven eosinophilic myeloid neoplasm, including those associated with rearrangements of PDGFRA and other recurrent molecular abnormalities), (2) lymphocytic variant HE/HES (presence of a clonal or phenotypically aberrant T-cell population that produces cytokines that drive the eosinophilia), (3) overlap HES (single-organ-restricted eosinophilic disorders and clinically defined eosinophilic syndromes that overlap in presentation with idiopathic HES; ie, eosinophilic gastrointestinal disorders, eosinophilic granulomatosis with polyangiitis), (4) associated HE/HES (in the context of a defined disorder, such as a helminth infection, neoplasm, immunodeficiency, or hypersensitivity reaction), (5) familial HE/HES (occurrence in >1 family member excluding associated HE/HES), and (6) idiopathic HE/HES (unknown cause and exclusion of other subtypes).
Lymphocytic variant HES (LHES) was first described in 1994 in a 30-year-old man with pruritus and cough and a large CD2+CD3–CD4+ T-cell population that produced interleukin (IL) 4 and IL-5 in response to stimulation.8 Since that time, other surface phenotypes have been described, and there have been several informative case series describing the clinical and laboratory findings of patients with LHES.9-11 Equally frequent in males and females, LHES most often presents with dermatologic manifestations. That said, any organ system can be involved, and some patients with asymptomatic HE have clonal aberrant T-cell populations indistinguishable from those with symptomatic LHES. Serum IgM, IgE, and serum and thymus and activation-regulated chemokine levels are elevated in most patients with LHES and can provide useful diagnostic clues.12 Whereas the gold standard for diagnosis of LHES is identification of a clonal and/or aberrant population of T cells producing type 2 cytokines, intracellular flow cytometry is not available at most centers, and the diagnosis most often relies on a compatible clinical picture and demonstration of a clonal and/or aberrant T-cell population in the peripheral blood. Expanded surface phenotyping is often necessary to demonstrate and/or confirm the aberrant population.12,13 It is important to recognize that the surface phenotype of the clonal population in LHES can be indistinguishable from that seen in T-cell malignancies (especially angioimmunoblastic T-cell lymphoma and cutaneous T-cell lymphoma).14 Thus, LHES is a diagnosis of exclusion. Moreover, progression of LHES to a lymphoid malignancy occurs in approximately 10% of patients, sometimes after many years of stable disease.15-17 Consequently, at a minimum, patients with LHES should undergo assessment for occult lymphoma at diagnosis and in the setting of an increase in size of the clonal T-cell population or development of resistance to previously effective therapy.
Approach to therapy
Despite the differences in definitions, the general approach to HE is very similar between World Health Organization and the consensus group (Figure 1). Since secondary causes of eosinophilia, such as helminth infection, typically require a different therapeutic approach, these should be considered early in the diagnostic process. If an underlying etiology is identified or highly suspected, specific treatment should be initiated. Primary (clonal/neoplastic) eosinophilia is also important to identify early due to prognostic and therapeutic implications.18 Finally, the presence and severity of clinical manifestations should be assessed as this will affect both the nature and urgency of therapeutic intervention. For example, careful monitoring without therapy may be appropriate for asymptomatic HE without evidence of end-organ involvement (hypereosinophilia of undetermined significance),19 whereas urgent intervention is needed in the context of myocarditis or thromboembolism.
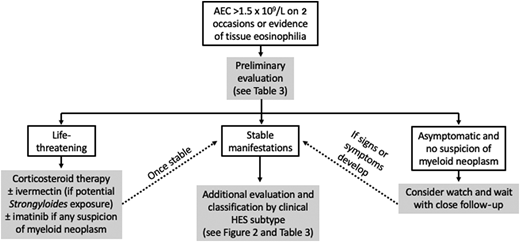
Prednisone remains the mainstay of therapy in the acute setting for severe and/or life-threatening manifestations of HES. If the eosinophilia does not dramatically decrease within 24 to 48 hours, additional therapy should be considered depending on the suspected clinical subtype (ie, imatinib for patients with clinical findings suggestive of a myeloid neoplasm, cyclophosphamide for patients with manifestations suggestive of eosinophilic granulomatosis with polyangiitis). The use of eosinophil-targeting biologics in the acute setting remains controversial but is supported by case reports and small series.20 Once the patient is stable, further evaluation should focus on the identification of the most likely clinical subtype (Table 3). With the exception of patients with myeloid HES,18 most patients with symptomatic HES respond rapidly to corticosteroid therapy, although toxicity and resistance limit the utility of this therapy in the long term.3 Conventional second-line agents, including hydroxyurea and interferon α, are fraught with similar issues but have advantages in select populations/clinical HES subtypes (Tables 3 and 4).
CLINICAL CASE (Continued)
Over the next 4 years, she remained relatively stable on interferon α therapy with partially controlled symptoms, AEC <1.0 to 2.0 × 109/L, but was unable to taper prednisone below 12.5 mg daily without significant worsening. Her clonal T-cell population increased to approximately 30% of total lymphocytes, prompting repeat CT scan and bone marrow, which were unchanged, and positron emission tomography (PET)/CT scan, which showed no evidence of lymphoma. She was enrolled on a phase 2 placebo-controlled trial of benralizumab.
Eosinophil-targeted therapies and HES
The availability of biologics targeting IL-5 (mepolizumab and reslizumab) and its receptor (benralizumab), all of which are approved by the US Food and Drug Administration for the treatment of asthma, has profoundly altered the approach to the treatment of idiopathic, lymphocytic, and overlap variants of HES. Whereas corticosteroids are still recommended as initial therapy in most cases, eosinophil-targeting biologics have shown excellent safety and efficacy profiles in the treatment of HES,21-23 leading to the recent approval of mepolizumab for HES and eosinophilic granulomatosis with polyangiitis and the initiation of phase 3 trials of benralizumab for the same indications. Of note, mepolizumab and reslizumab cause maturational arrest in the bone marrow with dramatic but incomplete reduction of blood and tissue eosinophilia. In contrast, benralizumab, an afucosylated monoclonal antibody to IL-5 receptor α, targets eosinophils, basophils, and their precursors for antibody-dependent cell-mediated cytotoxicity, resulting in complete (or near- complete) depletion in all tissues studied to date. Although definitive data are lacking, what little information is available concerning the efficacy of these agents in myeloid forms of HES is discouraging.22,24,25
Other agents that directly or indirectly decrease blood and tissue eosinophilia are in development for HES and/or approved for select eosinophilic indications (Table 4). These include lirentelimab (an afucosylated antibody to Siglec-8 that depletes eosinophils and basophils and inhibits mast cell activation),26 dupilumab (a monoclonal antibody to IL-4 receptor α approved for asthma, atopic dermatitis, chronic rhinosinusitis, and eosinophilic esophagitis that blocks IL-4 and IL-13 signaling and eotaxin-mediated tissue migration of eosinophils),27 and dexpramipexole (an orally available small molecule that causes maturational arrest and eosinophil depletion through an unknown mechanism).28
CLINICAL CASE (Continued)
After an initial response to benralizumab, interferon α was discontinued. Two weeks later, eosinophilia and severe symptoms returned. Benralizumab was discontinued, and she was started on prednisone 60 mg in addition to interferon α. She subsequently developed acute sensorineural hearing loss that resolved with cessation of interferon α and a prednisone burst. Cyclosporine was added. Due to persistent symptoms and inability to taper prednisone below 20 mg daily, she was enrolled on a phase 3 placebo-controlled trial of mepolizumab. Her symptoms improved, and she was able to taper prednisone to 9 mg daily, at which point she developed cough and shortness of breath requiring hospital admission. Evaluation was notable for ground-glass infiltrates, diffuse lymphadenopathy, and splenomegaly. Lymph node biopsy (approximately 10 years after her initial diagnosis) revealed angioimmunoblastic T-cell lymphoma. She was treated with CHOP-R (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate [Oncovin], prednisone, and rituximab) with transient response. Romidepsin was added, but she developed right-sided heart failure and died of respiratory failure.
Predictors of response to targeted therapy
There is currently little available information to guide the initial choice of biologic for a patient with HES. Although AEC has been shown to predict response to IL-5/IL-5 receptor targeting agents in patients with asthma, neither serum IL-5 levels nor eosinophil count at initiation of treatment were found to predict response to mepolizumab in the phase 3 trial in patients with PDGFRA-negative, corticosteroid-responsive HES,29 and neither historic peak nor baseline AEC predicted response to benralizumab in a phase 2 trial in patients with PDGFRA-negative, treatment-refractory HES.22 The only consistent finding across trials has been differences across clinical HES subtypes, with decreased response rates and/or increased relapse rates in patients with lymphocytic variant HES22,25,30-32 (Figure 2). That said, responses to the different biologics targeting the IL-5 axis are variable, and a lack of response to one agent does not preclude success with another.32
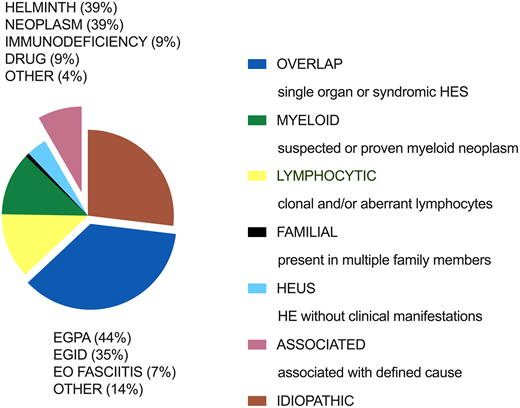
Clinical subtypes of hypereosinophilic syndrome: frequency distribution of 554 patients referred to the National Institutes of Health for unexplained eosinophilia. EGID, eosinophilic gastrointestinal disease; EGPA, eosinophilic granulomatosis and polyangiitis; EO FASCIITIS, eosinophilic fasciitis.
Clinical subtypes of hypereosinophilic syndrome: frequency distribution of 554 patients referred to the National Institutes of Health for unexplained eosinophilia. EGID, eosinophilic gastrointestinal disease; EGPA, eosinophilic granulomatosis and polyangiitis; EO FASCIITIS, eosinophilic fasciitis.
Potential risks of eosinophil depletion
Over the past 10 to 15 years, it has become increasingly apparent that eosinophils play an important role in homeostatic processes, including tissue remodeling, tumor surveillance, metabolic function, and immunoregulation.33 Although this has led to theoretical concerns about the effects of long-term therapy with biologics and other agents that significantly reduce eosinophils in blood and tissue, data to date suggest that eosinophil depletion in humans is safe.25,34-37 Importantly, there have been no reports of an increased incidence of malignancy or autoimmune disease related to eosinophil depletion, and even subtle immunologic consequences demonstrated in murine models, such as impaired vaccine responses, have not been replicated in human studies.22,34,38,39 This lack of significant toxicity is likely due, at least in part, to the redundancy and complexity of the human immune system, which raises potential concerns as the number of targeted therapies and biologics increases and the use of combination therapies becomes more common. It is also important to note that toxicities may be restricted to specific populations or clinical settings (ie, patients exposed to helminth infection, infected with coronavirus disease 2019, or at high risk of autoimmune disease), for which there are little to no prospective data to date.
Conclusions
Whereas eosinophilia is common in the general population, HES are a heterogeneous and complex group of rare disorders with clinical manifestations that span the range of medical subspecialties. Comprehensive clinical evaluation is necessary both to assess end-organ manifestations and determine the most likely etiology and/or clinical subtype of HES, as this information has important therapeutic and prognostic implications. Although corticosteroids continue to be first-line therapy in most situations, novel targeted therapies are rapidly replacing conventional cytotoxic and broad immunosuppressive agents as second-line agents of choice for the treatment of eosinophil-associated clinical manifestations. Despite the lack of safety signals to date, vigilance and prospective studies are needed to confirm the safety of these agents over the long term and to assess the impact of blocking multiple lineages and/or pathways on homeostatic processes.
Acknowledgment
This work was funded by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Conflict-of-interest disclosure
Amy D. Klion: no competing financial interests to declare.
Off-label drug use
Amy D. Klion: all of the drugs discussed, with the exception of imatinib and mepolizumab, are considered off-label for the treatment of hypereosinophilic syndromes.