Abstract
Chemoresistance remains a challenging clinical problem in the treatment of many lymphoma patients. Epigenetic derangements have been implicated in both intrinsic and acquired chemoresistance. Mutations in epigenetic processes shift entire networks of signaling pathways. They influence tumor suppressors, the DNA-damage response, cell-cycle regulators, and apoptosis. Epigenetic alterations have also been implicated in contributing to immune evasion. Although increased DNA methylation at CpG sites is the most widely studied alteration, increased histone methylation and decreased histone acetylation have also been implicated in stem-like characteristics and highly aggressive disease states as demonstrated in both preclinical models of lymphoma and patient studies. These changes are nonrandom, occur in clusters, and are observed across many lymphoma subtypes. Although caution must be taken when combining epigenetic therapies with other antineoplastic agents, epigenetic therapies have rarely induced clinical meaningful responses as single agents. Epigenetic priming of chemotherapy, targeted therapies, and immunotherapies in lymphoma patients may create opportunities to overcome resistance.
Learning Objectives
Understand general principles of chemoresistance in lymphoma: intrinsic and acquired
Recognize the role of epigenetics in contributing to chemoresistance and evasion of antitumor immunity
Gain knowledge regarding strategies to use epigenetic priming to overcome chemoresistance
Clinical case
In 2014, a 44-year-old woman presented with right flank and pelvic pain, 6 months of night sweats, and a 10-kilogram weight loss. Past medical history was significant for a bleeding gastric ulcer secondary to nonsteroidal anti-inflammatory drug use. Laboratory examination was notable for the following: hemoglobin, 7.0 g/dL; lactate dehydrogenase, 605 U/L; alkaline phosphatase, 209 U/L; γ glutamyl transferase, 16 U/L; albumin, 3.2 g/dL; and calcium, 10.6 mg/dL. Positron emission tomography/computed tomography scanning revealed hilar, para-aortic, mesenteric, and iliac adenopathy, and a renal mass, with involvement of bilateral psoas muscles and diffuse boney disease with standardized uptake values up to 21. Histologic evaluation of a psoas muscle biopsy revealed diffuse cellular infiltrate involving the skeletal muscle with large-sized cells positive for CD30, CD25, CD4, and CD5 and negative for anaplastic lymphoma kinase (ALK), consistent with ALK− anaplastic large cell lymphoma (ALCL). The patient was treated on the ECHELON-2 clinical trial and was randomized to receive either cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or brentuximab plus cyclophosphamide, doxorubicin, and prednisone.1 After 6 cycles, she attained a partial response and was consolidated with an autologous stem cell transplant. Posttransplant, she received 3000 cGy radiation for persistent disease in her humeral head. Six months later, the patient relapsed with cervical and pelvic lymphadenopathy and was treated on a clinical trial of combined epigenetic therapy with a novel antifolate, pralatrexate plus romidepsin.2 Following 2 months of treatment, a positron emission tomography/computed tomography scan confirmed a complete response. The patient received therapy for a total of 1 year. As of June 2020, she remains free from disease and has remained healthy without any untoward sequalae.
Introduction
The recognition of drug resistance dates back to the study of antibiotic effectiveness. Luria and Delbrück uncovered the emergence of antibiotic resistance in 1943. Many of their discoveries have been applied to cancer chemotherapy resistance.3 As Fox and Loeb have stated, “Cancers represent a microcosm of Darwinian evolution: tumor progression is a mutation-driven process that results from the adaptation of a heterogeneous cell population to different microenvironments through the preferential replication of the most suitable variants.”4
In patients with lymphoma, chemotherapy resistance occurs by 2 main mechanisms. Intrinsic drug resistance is defined by resistance to treatment without any prior exposure to chemotherapy. It is associated with reduced drug transport, drug breakdown, altered expression or engagement of the target, or specific intrinsic biological properties of the lymphoma, such as alterations in cell-cycle regulators, and DNA-damage repair mechanisms.5 Lymphomas harboring these attributes often have a dismal prognosis from the outset. One such example is the blastoid variant of mantle cell lymphoma characterized by cyclin D1 translocations and p53 mutations. Acquired drug resistance refers to the change in a susceptible tumor to one that has become resistant after repeated exposure to chemotherapy. This occurs via environmental factors that lead to genetic changes in the tumor, metabolic variations affecting the drug, modifications in the microenvironment and immune surveillance, and complex shifts of entire networks of signaling pathways. These events are often not related to specific chemotherapeutic drugs but rather are a result of intratumoral heterogeneity and random spontaneous events, which creates selective pressure for expansion of a clonal subpopulation. These mechanisms may occur alone, or more frequently together, which lowers the threshold for resistance to occur.6
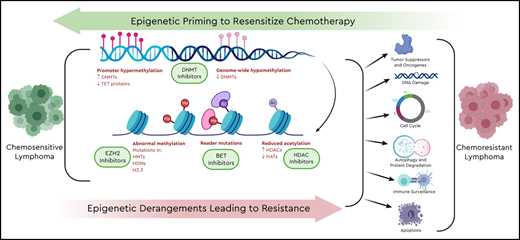
Strategies to overcome chemotherapy resistance and expand the spectrum of activity of both traditional chemotherapy and novel agents have been in development7 (Table 1). Epigenetic derangements have global effects, simultaneously influencing a greater set of pathways than direct genetic alterations of specific tumor-suppressor genes (Figure 1). Often, mutations in epigenetic processes do not occur in isolation, but rather simultaneously across multiple epigenetic controls, creating an opportunity to leverage epigenetic targeting as a means to overcome chemoresistance. This manuscript will build on the article “Harnessing lymphoma epigenetics to improve therapies” (see Green, in this book8 ). A better understanding of how targeting epigenetics have, and could be, used to overcome chemoresistance will hopefully spark innovation to improve outcomes for our patients with relapsed/refractory lymphoma.
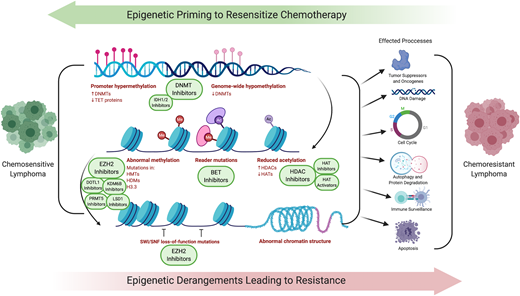
Epigenetic priming to resensitize chemotherapy. Derangements in multiple epigenetic processes lead to shifts in entire networks of signaling pathways, tumor suppressors, oncogenes, DNA-damage response, cell cycle, autophagy and protein degradation, and immune surveillance and apoptosis.
Epigenetic priming to resensitize chemotherapy. Derangements in multiple epigenetic processes lead to shifts in entire networks of signaling pathways, tumor suppressors, oncogenes, DNA-damage response, cell cycle, autophagy and protein degradation, and immune surveillance and apoptosis.
Epigenetic factors have been implicated in both intrinsic and acquired drug resistance
Epigenetic plasticity allows for a diversity of survival following exposure to chemotherapy by cooperating with somatic mutations to generate resistant subpopulations of lymphoma.9 Epigenetic polymorphisms, or epipolymorphisms demonstrate diversity both within and between lymphoma patient samples and has been found to evolve along with the development of a resistant clone.10 The term epiallele refers to a specific DNA methylation pattern at a discrete genetic locus. Following chemotherapy, tumor cells undergo significant epiallele alteration and, as resistant clones are selected, epipolymorphism diversity narrows.
DNA methylation has been the most widely studied epigenetic process contributing to chemoresistance. To study this, researchers created a phylogenetic tree to relate the level of methylation of lymphoma subtypes follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL) to patterning of normal B cells.9 They found that the “farther” the methylation pattern was from normal B cells, the worse the survival. This held true even between FL grades. Furthermore, abnormal methylation patterns were not random, but were directed at the promoter regions of key regulatory factors such as BCL6, EZH2, and MYC. Building on these observations, investigators studied methylation heterogeneity between DLBCL patients who maintained a durable treatment response to those who relapsed.11 They observed differentially hypermethylated regions enriched at promoter regions, specifically regulatory elements such as CTCF, a chromatin-binding factor that regulates the spread of DNA methylation through interactions with histone acetylases and deacetylases. In preclinical models of lymphoma,12 utilizing a mafosfamide-resistant cell line, investigators demonstrated that methylation was quantified at cytosine guanine dinucleotide (CpG) sites to identify differential methylation between the parental and resistant cell lines. The authors found that gene regions functionally enriched for DNA binding, transcription factor activity, and cell differentiation were marked with increased methylation. Additionally, members of the histone methyltransferase polycomb-repressive complex 2 were strongly enriched over repressed genes in the resistant cell line and the histone demethylase Uty was downregulated, contributing to chromatin condensation and decreased gene expression. The investigators found that the expression profiles of the resistant cells were similar to that of progenitor B cells, recapitulating stem-like characteristics. Similarly, in a study of ALCL, investigators found significant increases in CpG island methylation in the aggressive chemorefractory ALK− subtype as compared with the chemosensitive ALK+ subtype.13 Furthermore, samples from relapsed ALCL patients demonstrated even greater hypermethylation. Interestingly, the investigators found similar hypermethylation patterns of resistant ALCL when compared with the profiles of DLBCL, suggesting that these patterns may not be random events but are common across lymphoma subtypes. The hypermethylation signature of the more aggressive lymphomas reverted back to that of ALK+ ALCL following exposure to the DNA methyltransferase (DNMT) inhibitor 5-azacytidine. In a study designed to better understand the contribution of DNA methylation to chemoresistance and therapies to reverse these effects, investigators found that the SMAD1 member of the transforming growth factor β pathway is silenced via aberrant DNA methylation in chemoresistant DLBCL. Treatment with DNMT inhibitors reversed this phenomenon and sensitized cells to doxorubicin. These findings were translated to a clinical trial of azacitidine and rituximab plus CHOP chemotherapy in patients with DLCBL, which was well tolerated and led to a complete response in 11 of 12 patients.14
In addition to DNA methylation, epigenetic modification of histones has been recognized as an important contributor to chemoresistance. By directly leading to transcriptional silencing, enhancer of zeste homolog 2 (EZH2) and the polycomb-repressive complex 2 recruit DNMTs and DNA clusters over promoter regions known to be hypermethylated. In etoposide-resistant lymphoma cell lines, EZH2 expression has been linked to steering cells toward senescence rather than apoptosis. EZH2 inhibition induced sustained expression of p53 through modulation of MDM2-like p53 binding protein, while also upregulating p21 to overcome chemotherapy resistance.15 In preclinical chemoresistance models, EZH2-mediated silencing of Schlafen 11 (SLFN11) led to impaired DNA-damage repair. Recent studies have identified SLFN11, an inhibitor of DNA replication, as a genomic determinant of response to agents such as doxorubicin, platinum, and methotrexate.16 Treatment with EZH2 inhibitors restored SLFN11 expression and induced chemosensitivity in resistant models.17 Disruptor of telomeric silencing 1-like (DOTL1), an H3k79 methyltransferase, promotes DNA repair through homologous recombination. DOTL1 inhibition prevents the recruitment of p53-binding protein 1 (53BP1) to sites of double-strand breaks. DOLTL1 inhibitors thus could sensitize cancer cells to DNA-damaging agents.18,19 The DOTL1 inhibitor, pinometostat (EPZ-5676), is now in clinical trials. The histone lysine demethylase, KDM6B, was found to be correlated with reduced survival in DLBCL lymphoma patients receiving rituximab plus CHOP chemotherapy.20 Following evaluation of 181 patients, the survival rate was 48% for high expressers vs 71% for low expressers. When the investigators treated lymphoma cell lines with the KDM6B inhibitor GSK-J4, they found marked increase in apoptosis when combined with chemotherapy, suggesting chemosensitization. The authors also found that GSK-J4 influenced B-cell receptor signaling and B-cell lymphoma 6 (BCL6) expression, suggesting that these drivers of disease may be modified with this treatment.
Histone acetylation has been known to affect a broad array of cellular mechanisms ranging from apoptosis, DNA-damage response, cell cycle, autophagy, protein degradation, and immune response. Through modulation of these pathways, therapeutic targeting of histone acetylation, mainly through histone deacetylase (HDAC) inhibition, has been a strategy for overcoming resistance, with the chemoresistant T-cell lymphomas as the model for HDAC-inhibitor activity. Three HDAC inhibitors are approved by the US Food and Drug Administration (FDA) for relapsed/refractory T-cell lymphoma: vorinostat, romidepsin, and belinostat; chidamide is approved in China. Circling back to our case, romidepsin has demonstrated an overall response rate (ORR) of 25% to 39%.21,22 In the study published by Coiffier et al, there were 21 patients with ALK− ALCL, and although this disease entity is not marked by epigenetic derangements, 24% of those with ALCL achieved a response to therapy. Increased expression of p21 by HDAC inhibitors has been considered 1 potential mechanism of resensitizing chemoresistant lymphoma. This has been recognized across several lymphoma subtypes including DLBCL, mantle cell lymphoma, T-cell lymphoma, and Hodgkin lymphoma.23 In a study of primary refractory DLBCL patient samples, vorinostat inhibited cell viability, induced p21 expression, and downregulated cyclin-dependent kinase 2, leading to resensitization to chemotherapy.24 In rituximab-resistant DLBCL cell lines, entinostat led to decreased Bcl-XL levels and increased p21 and it potentiated the effects of cytarabine.25 Similarly, in Hodgkin lymphoma cell lines, vorinostat led to induction of p21 and downregulation of cyclin D2, producing synergistic cytotoxicity with cisplatin.26
BCLs treated with anti-CD20 monoclonal antibodies are frequently associated with downregulation of CD20, which breeds resistance to rituximab. Epigenetic modulation of the CD20-coding gene, MS4A1, has been shown to contribute to loss of CD20. Treatment with the HDAC6-selective inhibitor, ACY-1215, has been demonstrated to induce CD20 expression and sensitize cells to rituximab.27 This has also been demonstrated with entinostat and chidamide, suggesting a class effect.25,28
Epigenetics can also have effects on signaling pathways that are targetable with small molecules. One such example is targeting with the Bruton tyrosine kinase (BTK) inhibitor ibrutinib, in which resistance in mantle cell lymphoma has been recognized. Although mutations in the BTK-binding site are responsible for a subset of ibrutinib resistance, activation of alternative pathways leading to NF-κB signaling also play a role such as phosphatidylinositol 3-kinase (PI3K)-AKT activation.29,30 HDAC inhibitors have been demonstrated to enhance activity of PI3K inhibitors through modulation of the JAK/STAT pathway. As such, the dual HDAC/PI3K inhibitor CUDC-907 has demonstrated activity in BTK-resistant mantle cell lymphoma and DLBCL preclinical models.
HDAC inhibitors have been implicated in inducing both mitochondrial and death receptor apoptotic pathways. Investigators studied a panel of etoposide-resistant lymphoma cell lines noted to have mutant BCL2.31 Treatment with romidepsin led to acetylation and inactivation of heat shock protein 90, allowing for the upregulation of Bcl2-like protein 11. Although this effect is based on posttranslational rather than epigenetic modification, acetylation of heat shock protein 90 by HDAC inhibitors increased sensitivity to chemotherapy. Induction of Bcl2-like protein 11 has been noted across multiple lymphoma subtypes following exposure to HDAC inhibitors. As such, combination with BCL2 inhibitors such as venetoclax has demonstrated promising results.32
Posttranslational modifications induced by epigenetic drugs have the ability to influence many pathways known to contribute to chemoresistance. One such example is impaired p53 activity. Although there are many mechanisms responsible for reduced activity, acetylation of p53 activates the tumor suppressor and protects it from degradation.33 Treatment with sirtuin inhibitors, class III HDAC inhibitors, can induce acetylation of p53. HDAC inhibitors have been known to influence the effects of master regulators and key oncogenes such as BCL6.34,35 Our laboratory demonstrated synergy between romidepsin and niacinamide, a sirtuin inhibitor, through enforcing acetylation of the oncogene BCL6, abrogating its effects, and acetylation of p53 activating the tumor suppressor.31 Romidepsin has been demonstrated to downregulate BCL6 and influence its target genes such as cyclin D2 and PRDM1. This in turn has led to synergistic targeting of MYC with the bromodomain and extraterminal domain (BET) inhibitor JQ1.
Intersection of epigenetics and immunotherapy
Immune evasion has been increasingly recognized as a mechanism of disease progression in lymphomas (Figure 2). Epigenetic regulation of immune surveillance in the microenvironment is an area of intense study (Table 2). Acquired chemoresistance has been linked to increased expression of programmed cell death 1 (PD-1) and programmed death ligand 1 (PD-L1), which have been demonstrated to be under epigenetic control.36 De novo DNA methylation of promoter regions of Pdcd1 fix T cells in an exhausted state. Although this may be reversed with checkpoint blockade, when treatment is withdrawn, T cells revert to an exhausted state. Treatment with DNMT inhibitors has proven complimentary to checkpoint blockade, allowing for rejuvenation of the T-cell response.37-39 Additionally, histone acetylation of both the PD-L1 and PD-1 promoter regions is implicated in PD-L1 expression. Accordingly, treatment with HDAC inhibitors has been demonstrated to increase expression in both.40 Many of these studies have been performed in preclinical models of solid-organ malignancies or infection; however, their findings are now being explored in models of lymphoma and have rapidly been translated to clinical trials for relapsed/refractory lymphoma (Table 2).
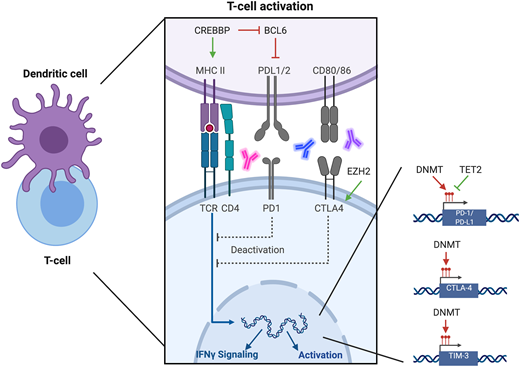
Intersection of epigenetics and immune surveillance. Regulation of immune surveillance is controlled in part by epigenetic operations. The histone acetyltransferase enzyme CREBBP activates expression of MHCII, which allows for engagement between the antigen-presenting cell and the T cell. CREBBP also abrogates BCL6, which in turn abrogates PD-L1 expression. These effects have been reversed with HDAC inhibitors. EZH2 is associated with increased CTLA4 activity therefore EZH2 inhibitors have combined favorably with CLTA4 inhibitors. Expression of PD-1, PD-L1, CTLA4, and TIM-3 is controlled by promotor region methylation; therefore, their expression may be modulated by DNMT inhibitors.
Intersection of epigenetics and immune surveillance. Regulation of immune surveillance is controlled in part by epigenetic operations. The histone acetyltransferase enzyme CREBBP activates expression of MHCII, which allows for engagement between the antigen-presenting cell and the T cell. CREBBP also abrogates BCL6, which in turn abrogates PD-L1 expression. These effects have been reversed with HDAC inhibitors. EZH2 is associated with increased CTLA4 activity therefore EZH2 inhibitors have combined favorably with CLTA4 inhibitors. Expression of PD-1, PD-L1, CTLA4, and TIM-3 is controlled by promotor region methylation; therefore, their expression may be modulated by DNMT inhibitors.
Germinal center BCLs, DLBCL and FL, are not overtly responsive to checkpoint inhibition partly due to immune evasion. These entities are marked by activating mutations of EZH2 and inactivating mutations in acetyltransferases EP300 and CREBBP. EZH2 suppresses activity in T cells and regulates chemokines, leading to reduced antitumor immunity.41 Investigators found that treatment with EZH2 inhibitor CPI-1205, reprogrammed T-cell antitumor immunity, suppressed regulatory T cells, and improved responses to anti-CTLA4 therapy. Derangements of both EZH2 and acetyltransferases have been implicated in reduced major histocompatibility complex (MHC) expression and tumor-infiltrating lymphocytes. Treatment with EZH2 inhibitors leads to increased expression of the class II MHC transactivator enhancer, restoring MHCII expression. In addition, programmed cell death 1 and CTLA4 were increased following EZH2 inhibition.42 CREBBP enhances MHCII expression and abrogates BCL6 inhibition of PD-L1. Mutations leading to CREB-binding protein (CREBBP) insufficiency have been linked to impaired B-cell to T-cell engagement and reduced T-cell expansion, activation, and antitumor immunity. Targeting this biology with HDAC3 inhibitors demonstrated a reversal of this phenomenon in preclinical models of DLBCL, resulting in synergistic cytotoxicity with PD-L1 inhibitors.43 In a genomic analysis of microdissected Hodgkin Reed-Sternberg cells from 12 patients with primary refractory disease, 4 of 12 cases had mutations in EP300 or CREBBP suggesting that this could play a role in reduced antitumor immunity.44 This implies that priming with epigenetic modulators could potentiate checkpoint blockade in diseases marked by impaired immune surveillance (Table 2).
In peripheral T-cell lymphoma (PTCL), specifically those of follicular helper cell of origin such as angioimmunoblastic T-cell lymphoma, mutations in Tet methylcytosine dioxygenase 2 (TET2) are often observed.45 TET2 is a dioxygenase that converts 5-methylcytosine to 5-carboxylcytosine. In addition, TET2 enzymatic activity is inactivated by the oncometabolite hydroxyglutarate generated by mutated isocitrate dehydrogenase 1/2 detected in up to 45% of angioimmunoblastic T-cell lymphoma patients.46,47 Reduced TET2-mediated demethylation of PD-L1 promoter regions, chemokines, and the interferon γ pathway contribute to T-cell exhaustion.48 These effects have been demonstrated to be reversed by treatment with DNMT inhibitors.49,50 Adult T-cell leukemia/lymphoma is a highly aggressive disease associated with both intrinsic and acquired chemoresistance. Investigators surveyed adult T-cell leukemia/lymphoma patient samples and found that 25 of 58 had increased expression of T-cell immunoglobulin and mucin-domain containing 3 (TIM-3), which was associated with chemoresistance.51 TIM-3 is expressed on immune cells such as macrophages, dendritic cells, natural killer cells, and T cells. Expression of TIM-3 regulates the immune response through downregulation of the interferon pathway leading to T-cell exhaustion. Expression of TIM-3 and its ligand, LGALS9, is known to be regulated by CpG island methylation within the promoter region, suggesting that expression could be regulated with DNMT inhibitors.52
Rewiring pathways driving chemotherapy resistance
There have been a multitude of studies evaluating combined epigenetic targeting for the treatment of lymphoma because aggressive lymphomas often have multiple epigenetic derangements and single-agent therapy has not proven clinically effective in many cases (Table 3). In preclinical studies of DLBCL, both vorinostat and decitabine induced cytotoxicity in chemoresistant cell lines and the combination was synergistic.53 This has been demonstrated across other HDAC and DNMT inhibitor combinations.54 The investigators translated these findings into a phase 1b study of relapsed refractory DLBCL patients and treated them with vorinostat and azacytidine; however, the study was closed early secondary to hematologic toxicity and disease progression. Interestingly, 7 of 18 patients went on to further treatment, 2 of whom achieved a complete response; 3 achieved clinical benefit from their subsequent therapies with responses ranging from 79 to 825 days. In a similar effort, investigators treated patients with relapsed DLCBL with the chidamide combined with decitabine then sequentially with rituximab plus gemcitabine and oxaliplatin.55 All 13 patients enrolled achieved disease control with 3 achieving a complete remission. Not surprisingly, grade 3-4 hematologic toxicities were frequent, as were gastrointestinal side effects, mucositis, pyrexia, and liver abnormalities. Finding the appropriate dosing schedule and patient population to best benefit from combined epigenetic therapy will be necessary for successful application. For example, our team has demonstrated synergy between EZH2 and HDAC inhibitors only in cell lines with deranged EZH2 expression or activity, and not in those with normal EZH2 expression.56 Our team evaluated the merits of the combination of oral azacitidine and romidepsin, for which the majority of responses were seen in T-cell lymphoma patients. Interestingly, among the 3 patients with germinal center–derived BCL, disease control was achieved in 1 patient with stable disease, 1 patient with a partial response, and 1 with a complete remission. Although the numbers with germinal center B-lymphomas are exceedingly small, this disease entity is marked by epigenetic derangements.50
Conclusion
Resistance to both chemotherapy and targeted small molecules continues to limit our ability to cure many with lymphoma. Epigenetic modulating drugs may be used in concert with other epigenetic targeting agents, or small molecules with the intention of rewiring pathways circumnavigating resistance. They may be used to enhance the effects of checkpoint inhibitors. Caution must be taken in efforts to combine these therapies as toxicity could limit their usefulness. History has taught us that epigenetic drugs are unlikely to have clinical meaningful effects alone. Recognizing patterns of epigenetic derangements in specific disease entities could create an opportunity for precision medicine that will allow epigenetic therapy to be combined effectively. One of their greatest impacts may be as primers to chemotherapy or immunotherapy to overcome resistance.
Acknowledgments
Funding/grant support was provided by the Irving Scholar Program, The Irving Institute for Clinical and Translational Research, Columbia University Irving Medical Center. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA222931. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Correspondence
Jennifer E. Amengual, Columbia University Medical Center, William Black Building, Room 905, 650 West 168th Street, New York, NY, 10035; e-mail: jea2149@columbia.edu.