
Abstract
Myelodysplastic syndromes (MDS) are a heterogeneous group of disorders characterized by clonal hematopoiesis with a propensity to evolve into acute myeloid leukemia. MDS presenting in children and young adults is associated with features clinically and biologically distinct from MDS arising in older adults. MDS presenting in children and young adults is associated with a higher likelihood of an underlying genetic predisposition; however, genetic predisposition is increasingly recognized in a subset of older adults. The diagnosis of a genetic predisposition to MDS informs clinical care and treatment selection. Early diagnosis allows a tailored approach to management and surveillance. Genetic testing now offers a powerful diagnostic approach but also poses new challenges and caveats. Clinical expertise in these disorders together with scientific expertise regarding the affected genes is essential for diagnosis. Understanding the basic mechanisms of genetic predisposition to myeloid malignancies may inform surveillance strategies and lead to novel therapies. The cases presented in this article illustrate challenges to the diagnosis of germline genetic predisposition to MDS and how the diagnosis affects clinical management and treatment.
Learning Objectives
Identify the clinical implications of genetic predisposition to MDS, including changes to medical management and family counseling
Implement strategies to identify patients with genetic predisposition to MDS
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of disorders characterized by clonal hematopoiesis with a propensity to progress to acute myeloid leukemia (AML).1 The ineffective hematopoiesis of MDS typically presents with peripheral blood cytopenia and bone marrow dysplasia. Although MDS in older adults typically presents with age-associated acquisition of somatic mutations driving clonal evolution, children and young adults are more likely to harbor a germline genetic condition leading to MDS.2
MDS due to germline predisposition is classified as a separate entity under the 2016 World Health Organization system.3 The germline disorders associated with MDS predisposition are heterogeneous and rare individually; however, collectively, they account for 4% to 15% of MDS cases.4-11 This frequency is likely higher, because many patients lack characteristic physical stigmata or may not have undergone a comprehensive evaluation for an underlying MDS predisposition syndrome.12 Ongoing identification of additional causative genes and mutations and improvement in testing methodologies continue to increase recognition of these disorders. Increasing clinician awareness of MDS genetic predisposition disorders and their phenotypic variability is critical to inform tailored optimal clinical care. Investigation of disease biology and the molecular mechanisms driving clonal evolution to myeloid malignancies is ongoing to improve surveillance strategies and to develop novel therapies.13 The cases described in this article illustrate the challenges to the diagnosis of germline genetic predisposition to MDS and how these diagnoses profoundly affect clinical management and treatment.
Clinical case 1
A 26-year-old woman presents for consultation regarding MDS. She was treated elsewhere 10 years ago for high-risk hypodiploid pre–B-cell acute lymphoblastic leukemia (ALL) and achieved remission with chemotherapy. She relapsed with the same pre–B-cell ALL 8 years later and underwent reinduction chemotherapy.
She experienced multiple prolonged chemotherapy delays due to persistent thrombocytopenia during the maintenance phase of treatment. A bone marrow examination performed for further evaluation showed a hypocellular marrow with multilineage dysplasia and a complex karyotype. Fluorescence in situ hybridization (FISH) was notable for monosomy 7, del5q, and monosomy 20.
She has 2 healthy siblings, and her brother is a 10/10 HLA match. Her father is alive and healthy (age 52 years). Her mother died with breast cancer at age 33 years and had undergone BRCA1/2 testing, the result of which was negative. A maternal aunt and grandmother died of breast cancer; a maternal uncle had a history of adrenal cortical carcinoma and pancreatic cancer; a maternal aunt was treated for melanoma; and a maternal cousin was treated for a brain tumor.
The patient is in the 40th percentile for height with no dysmorphic facial features. She has normal dentition and oral mucosa; normal cardiac and respiratory examination results; no abdominal masses or hepatosplenomegaly; normal extremities, radial pulses, and nails; and a normal neurological examination result. She has no rashes or skin findings. Complete blood counts (CBCs) before initial leukemia diagnosis were notable for no cytopenia or macrocytosis. A genetic panel for cancer predisposition had been sent from an outside hospital to a commercial laboratory several years earlier, and a comment in the clinical summary from the referring physician mentioned that the result of genetic testing for TP53 mutations was negative.
Initial evaluation
An approach to the evaluation of inherited predisposition to myeloid malignancy is outlined in Figure 1. MDS predisposition syndromes and associated genes are listed in Table 1, and clinical features flagging a potential MDS predisposition disorder are listed in Table 2.14-19 An initial history for a patient with MDS includes a thorough evaluation for prior cytopenia; medical comorbidities; prior malignancies; and response to therapy, including assessment of excessive toxicity to chemotherapy, hematopoietic stem cell transplant (HSCT) conditioning, or radiation therapy.
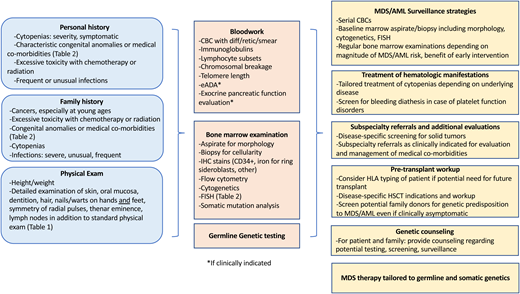
Workup of a patient with suspected MDS predisposition syndrome and management implications. eADA, erythrocyte adenosine deaminase; HSCT, hematopoietic stem cell transplant; IHC, immunohistochemistry.
Workup of a patient with suspected MDS predisposition syndrome and management implications. eADA, erythrocyte adenosine deaminase; HSCT, hematopoietic stem cell transplant; IHC, immunohistochemistry.
A detailed 3-generation family pedigree taken for this purpose includes specific questions regarding cytopenia or bleeding phenotypes, frequent or unusual infections, early deaths or miscarriages, hematologic malignancies and solid tumors, any toxicities with therapy, response to treatment, congenital anomalies, and other clinical stigmata of genetic MDS predisposition syndromes (Table 2). Many disorders are associated with intrauterine growth retardation, so birth weight and prenatal history should be ascertained. It is useful to revisit and update the family history at subsequent visits because complications may develop over time, and it allows families to glean additional information from their relatives.
A physical examination starts with assessment of both linear growth over time in children and weight percentiles/body mass index. A careful evaluation for physical findings suggestive of a genetic disorder is essential; however, disorders classically considered syndromic may present with only MDS and may lack any additional apparent stigmata. Several disorders associated with MDS are associated with classical facial features, such as Fanconi anemia (FA), Diamond-Blackfan anemia (DBA), and Bloom syndrome. Similarly, some disorders, such as FA and DBA, can present with thumb and radial ray anomalies that may manifest with subtle findings, such as a hypoplastic thenar eminence or asymmetry of the radial pulses. Examination of skin, hair, dentition, and nails can identify characteristic changes in dyskeratosis congenita (DC)/telomere biology disorders (TBDs) and Werner syndrome. Such abnormalities due to TBDs have masqueraded as chronic graft-versus-host disease. Nail abnormalities in TBDs may present with thin and fragile nails; thickened, ridged, or discolored nails; or poor nail growth. Nail abnormalities may be erroneously attributed to chronic fungal infection unresponsive to treatment. Nail abnormalities may lack uniformity, with only a single abnormal nail present. Tactful inquiry regarding application of artificial nails or hair dyes is essential to avoid missing important indicators of a genetic disorder. Nijmegen breakage syndrome, neurofibromatosis 1, FA, TBD, and Bloom syndrome can present with hypo- or hyperpigmented skin lesions. Warts or molluscum may signal an underlying immunodeficiency such as that seen with GATA2 deficiency or ligase IV syndrome. Skeletal anomalies, microcephaly, and genitourinary anomalies can be present in many disorders. Examining both upper and lower extremities is useful because nail or digit abnormalities or dermatologic findings or lymphedema may be visible only on upper or lower extremities (Table 2). Examining family members’ hands, nails, and so forth during a clinic visit can also provide useful clues.
Prior CBCs are informative. Assessment includes evaluation for macrocytosis and any cytopenia, including enumeration of neutrophils, monocytes, lymphocytes, and reticulocytes. Review of the blood smear may reveal characteristic dysplasia. An immunologic screen including immunoglobulins and lymphocyte subsets (including B cells and natural killer cells) is helpful to flag genetic MDS predisposition disorders associated with immunologic abnormalities. Bone marrow analyses are outlined in Figure 1, and they include cytogenetic assessment and molecular studies.
Screening for exocrine pancreatic dysfunction is helpful in the workup of Shwachman-Diamond syndrome (SDS) and includes a history of steatorrhea, which is often most pronounced in infancy/early childhood; serum trypsinogen for those less than 3 years of age; and pancreatic isoamylase for patients greater than 3 years of age. Testing for DC includes telomere lengths of lymphocyte subsets. Testing for FA includes chromosomal breakage studies in response to diepoxybutane and mitomycin C (Figure 1). DC and FA are associated with high transplant regimen–related toxicities and require modified transplant conditioning regimens.20-22 Of note, chromosomal breakage may be elevated at baseline in patients who have received prior chemotherapy, but increased breakage with diepoxybutane and mitomycin C is a classic feature of FA. Similar patterns of chromosomal breakage are seen in Nijmegen breakage syndrome. Somatic mosaicism may result in a false-negative chromosomal breakage test result for FA, so testing of fibroblasts grown from a skin punch biopsy may be considered. Telomere length analysis may be challenging in cases of prior chemotherapy, but normal telomere length testing can be reassuring evidence against an inherited telomeropathy.
In clinical case 1, the patient’s personal history of hypodiploid ALL followed by MDS, as well as her family history of multiple characteristic solid tumors, including early-onset breast cancer, adrenal cortical carcinoma, brain tumors, pancreatic tumors, and melanoma, meets Chompret criteria for Li-Fraumeni syndrome, which is associated with germline TP53 mutations.23,24 The primary laboratory report from prior genetic testing were requested and reviewed. The report read, “Result: negative, no clinically significant mutation identified.” Examination of the primary genetic report revealed deep in the text a variant in TP53, c.389T>A (p.Leu130His), which had been labeled a variant of uncertain significance (VUS) in the laboratory report in 2014.
Genetic testing
Before sending genetic testing, genetic counseling is recommended to review testing options, range of possible results, and possible implications for the family. Expertise in both the genetic MDS disorders and genetic counseling is essential and may be provided in partnership between an expert hematologist and genetic counselor experienced with these disorders. Genetic test results typically take weeks to months to return; thus, it may be challenging to send for these tests in clinically urgent cases when patients have already developed MDS. For this reason, early genetic evaluation before the development of clinical complications is advantageous. It is important to note the various testing platforms available, including Sanger sequencing and next-generation sequencing (NGS) of single genes, NGS panels with or without ability to detect duplications and deletions, whole-exome sequencing, and whole-genome sequencing. Each MDS predisposition panel offers different subsets of genes, so knowledge of included and excluded genes for any given panel is essential. Additional genes remain to be discovered for multigenic disorders such DC and FA. Clinical considerations of genetic testing are covered in recent reviews.25-27
Analysis of a VUS
Clinical case 1 highlights the complexity of genetic testing. Medical expertise in the disease and scientific expertise in the genes and mutations are required. Direct examination of primary genetic reports is essential.
In some cases, evaluation of a mutation is straightforward if the mutation has previously been reported to be pathogenic, particularly if the variant tracks with the phenotype and inheritance pattern in multiple unrelated families and functional studies confirm that the variant affects a gene function resulting in disease pathogenesis. Alternatively, the variant may be a known benign polymorphism. However, in other cases, a variant may be labeled a VUS. Considerations in evaluating a VUS are outlined in Figure 2.
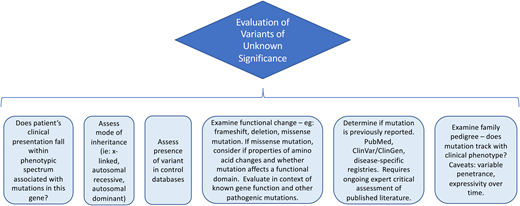
An initial useful screen in the workup of a VUS examines the allele frequency in control databases. In clinical case 1, the patient’s mutation was absent from control databases. However, it is critical to note that control databases can include patients with a mild or clinically cryptic or delayed phenotypic spectrum and that recessive variants for rare diseases may be present in the general population.
Next, it can be helpful to assess the functional implication of the variant. In clinical case 1, the variant is located in the highly conserved DNA-binding domain of TP53, which raises the suspicion that this might be a pathogenic mutation. Understanding whether the disease in question is associated with activating mutations or loss of protein function is also critical for predicting pathogenicity. Disease-specific databases can be essential for curation; in this case, this variant was reported in a database assessing functional effects of TP53 variants. Variants are constantly being reported, curated, and updated, and thus a VUS requires reanalysis over time. Consultation with an expert in the disease and gene of interest is recommended.
Impact on treatment
In clinical case 1, identifying a pathogenic variant affected multiple aspects of the patient’s care (see Figure 1).28,29 The patient was screened for solid tumors before proceeding to transplant. Patients with Li-Fraumeni syndrome are sensitive to radiation and are at high risk of secondary malignancies; the transplant conditioning regimen should be tailored to minimize toxicity. In addition, the patient’s matched sibling donor could be screened for the pathogenic variant to ensure that he was unaffected. Genetic counseling should be offered to the rest of the family and is critical for discussions regarding surveillance and family planning.
Timing of MDS predisposition evaluation
The benefits of assessing for genetic predisposition to MDS/leukemia must be weighed against the risk of delay to transplant. Initiating the evaluation as early as possible is helpful for optimal patient care. Outcomes are best if transplant is performed before disease advances.30 Deferring genetic testing until the patient appears ill might delay urgently needed therapy.
Additional considerations
In cases with a high suspicion for an inherited cancer predisposition syndrome, it is essential to carefully evaluate any potential related donors, even if the results of the patient’s workup are negative. Donor evaluation includes a medical history, physical examination, and screening blood tests (listed in Figure 1). Some providers perform a bone marrow evaluation that includes morphology, cellularity assessment, karyotype, and MDS FISH as part of a related-donor evaluation for pediatric MDS, given high rates of inherited predisposition.
Clinical case 2
A previously healthy 15-year-old boy presented with fatigue, pallor, and pancytopenia, as well as a hypocellular marrow with multilineage dysplasia, 10% blasts, and a monosomy 7 clone. He was diagnosed with MDS. As part of his workup, an NGS panel for somatic mutations in hematologic malignancies was sent, which did not reveal any mutations. Because monosomy 7 is associated with GATA2 deficiency, it was confirmed that the NGS panel included GATA2, and the result was negative.
Plans were made for definitive treatment with a matched sibling donor transplant. One of his 4 siblings was a full HLA match. The sibling was healthy other than a history of eczema and ear infections requiring tympanostomy tubes.
Characteristic cytogenetic and molecular findings
Certain cytogenetic changes are frequently seen in MDS predisposition syndromes. Monosomy 7 and del7q are especially frequently seen in genetic predisposition conditions such as DC, FA, GATA2 disorders, SAMD9/SAMD9L, GATA2 disorders, SDS, and severe congenital neutropenia (Table 3).31,32 Physical examination findings and laboratory findings associated with these conditions are included in Table 2.
Familiarity with the most frequent cytogenetic clones and somatic mutations arising in inherited bone marrow failure syndromes can be helpful in performing targeted workup. Certain MDS predisposition syndromes have characteristic somatic mutations, such as DNMT3A mutations in MBD4 (Table 3).33 Cytogenetic alterations include +1q, 3q amplification, +13q in FA, and del20q and iso(7)q in SDS, among others (Table 3).15 The definitive screening and diagnostic testing approach for SAMD9/SAMD9L and GATA2 is genetic testing, whereas functional tests are available to screen for DC and FA (see clinical case 1).
Somatic reversion
Interestingly, del7q and monosomy 7 arise in SAMD9/SAMD9L as a result of selective pressures to circumvent the repression of hematopoiesis imposed by the activating mutations in SAMD9/SAMD9L genes located at the 7q22 locus. Additional molecular mechanisms to inactivate the mutant SAMD9 and SAMD9L allele include acquisition of a loss-of-function mutation in cis to the pathogenic mutation or uniparental disomy resulting in copy neutral loss of heterozygosity to remove the mutant allele.7,34-36 Germline mutations can be difficult to detect in the peripheral blood because of this frequent somatic correction and may only be present in low variant allele frequencies in the blood and marrow.34,35,37 Therefore, if there is high suspicion for these disorders, testing of nonhematopoietic tissue may be required for diagnosis.38
Signs and symptoms of MIRAGE syndrome (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy; caused by SAMD9 mutations) include infections, growth restriction, adrenal insufficiency, genital phenotypes, and enteropathy. However, the phenotypic spectrum is broad, and features of MIRAGE other than MDS may be absent. Similarly, somatic reversion in peripheral blood has been described in other inherited marrow failure syndromes, such as FA and DC, which can make diagnosis challenging, and in cases in which there is a high suspicion for the disorder, screening nonhematopoietic tissue can lead to the diagnosis.39,40
Chromosomal breakage studies were sent, and the results were negative. Telomere lengths were within normal range for total lymphocytes and granulocytes, but tests could not be performed on lymphocyte subsets, because the absolute numbers of lymphocytes were too low. A more detailed family history was taken, and parental CBCs were reviewed. The patient’s father was noted to have mild thrombocytopenia with a platelet count in the 130,000/μL range, and mild macrocytosis was detected with a mean corpuscular volume ranging between 90 and 95 fL in his prior CBCs. The donor workup was notable for an absolute monocyte count of 80 and low absolute natural killer cell numbers. Given the suspicion for germline GATA2 deficiency, Sanger sequencing was performed, and the results were returned with an intron 5 mutation in GATA2 (c.1017+572C>T) in the patient, brother, and father.41
Genetic testing considerations
As illustrated in clinical case 2, GATA2 can be somatically mutated in hematologic malignancies and is frequently included in many of the somatic mutation panels. However, many tests do not fully cover the entire gene. Coverage of pathogenic noncoding regions and analysis of non–protein-coding genes must also be considered. Thus, it is critical to know the coverage of the gene before relying on a specific test for screening of germline mutations. Not all mutations are easily detected by sequencing of exons. Small deletions and duplications or entire gene deletions can be difficult to detect without additional copy number analysis. In GATA2 as well as in other genes, there can be noncoding mutations that are pathogenic. Intron 5 in GATA2 contains a highly conserved region that is critical for enhancer activity, and mutations in this region are associated with GATA2 haploinsufficiency.41
Germline versus somatic mutations
Hematopoietic tissue can be affected by somatic mutations in the presence or absence of MDS or leukemia. Of note, many genes may be mutated either constitutionally or somatically (eg, GATA2, RUNX1, ETV6, and TP53), with profoundly different clinical implications.42 Although skin fibroblasts are generally considered the current gold standard for clinical testing of germline variants for MDS predisposition, this is a rapidly advancing field. Care is needed to select the most appropriate test up front. Although testing of peripheral blood DNA may be more expedient and most practical in the short term, it can lead to false-positive results (due to identifying somatic mutations) and false-negative findings (due to somatic reversion). Therefore, testing skin fibroblasts or other nonhematopoietic tissue may be required to distinguish germline from somatic mutations.38 Because of the extended time required for skin fibroblast culture, if there is a time constraint on definitive diagnosis, the process for skin fibroblast testing should be initiated as soon as possible. Testing family members may also be informative.
Clinical case 3
A 33-year-old woman is referred to a hematology clinic by her gynecologist for neutropenia (600 cells/μL) and thrombocytopenia (120,000/μL) found in a screening CBC. Her hemoglobin concentration was 11 g/dL, and her mean corpuscular volume was 90 fL.
The patient has a history of frequent pneumonia and acute otitis media in childhood, poor dentition, and gingivitis. On review of systems, it was noted that she had had steatorrhea as an infant that resolved in early childhood. She stood 154.94 cm tall. Her mother’s height was 175.26 cm, and her father’s was 182.88 cm. Her family history was negative for leukemia, solid tumors, frequent infections, cytopenia, failure to thrive, and steatorrhea.
The patient’s hematologist noticed that she had easy bruising. Her prothrombin time was mildly elevated, and her vitamin K level was low. She sent a pancreatic isoamylase level, which was low. Sanger sequencing on SBDS was sent and was returned with homozygous splice site mutations (c.258+2T>C, IVS2+2T>C).
Importance of pre-MDS diagnosis
Patients with milder phenotypes may be unrecognized until adulthood. Patients may have worsening of cytopenia during pregnancy.43 Recent studies demonstrate that inherited predisposition to MDS is underdiagnosed.7,44 Diagnosing a patient with an MDS predisposition syndrome before development of MDS or leukemia allows surveillance to allow intervention before the development of malignancy (Figure 1). Survival after progression to MDS or AML is poor for patients with many of these disorders, so surveillance allows transplant before disease progression. Therapy is limited by the increased toxicities associated with chemotherapy and radiation for many of these disorders.
Patients with SDS who received transplants for MDS had poor outcomes.4 Data from the SDS Registry suggest an association between leukemia surveillance and superior survival (E.F. and A.S., unpublished data), and further studies are ongoing. Enlisting the expertise of a hematopathologist experienced with MDS predisposition syndromes is critical to distinguish dysmorphologies and dysplasias associated with MDS predisposition disorders at baseline from MDS.
Timely diagnosis also allows screening of potential family members and tailoring of the transplant regimen to minimize toxicities related to underlying disease. Patients can also undergo early screening for medical comorbidities associated with their diagnosis (ie, screening for immunodeficiencies, bleeding disorders, endocrinopathies, pulmonary or hepatic complications, malformations). It is also critical to know the solid tumors that are associated with specific disorders and to note that disorders associated with immunodeficiencies can be associated with infection-related cancers such as human papillomavirus–related cancers (Table 2). Nontransplant disease-specific treatment of cytopenia may include androgens for FA and DC and steroids for DBA.
Conclusions
Maintaining a high index of suspicion for inherited MDS predisposition disorders is essential, especially because the phenotypes can be variable. Timely workup with a combination of functional and genetic testing allows accurate early diagnosis to inform a tailored approach to medical management and leukemia surveillance. Genetic testing allows previously unprecedented diagnostic precision but also poses new challenges. Partnering with a hematologist with expertise in MDS predisposition syndromes for diagnosis and medical management can be helpful.
Understanding of the genomic landscape of MDS in de novo MDS and of MDS arising from germline disorders is rapidly increasing.45,46 Advances in the underlying biology of MDS predisposition syndromes and clonal evolution may allow the development of more effective surveillance strategies. These in turn can lead to improved risk stratification algorithms for deciding when to move toward HSCT.
Acknowledgments
The authors regret that, owing to space limitations, inclusion of all publications on this topic was not possible. The authors apologize for any omissions and refer the interested reader to additional primary references in cited reviews.
This work was supported in part by National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases grant R24 DK099808 (A.S.) and NIH grant T32 HL007574-36 (E.F.).
Correspondence
Akiko Shimamura, Boston Children’s Hospital, 300 Longwood Ave, Karp 8210, Boston, MA 02115; e-mail: akiko.shimamura@childrens.harvard.edu.
