Abstract
Treatment of hemophagocytic lymphohistiocytosis (HLH) has been developed primarily in pediatric centers, where familial HLH (FHL) is the leading cause of HLH in newborns and toddlers. The Histiocyte Society Study Group for HLH developed the HLH-94 and HLH-2004 treatment protocols, and these are frequently also used by centers treating HLH in adults (aHLH). These protocols contain etoposide, dexamethasone, and cyclosporine A; these agents all have strong activity against proliferation of cytotoxic T/NK-cells and macrophages, as well as inhibitory activity against the cytokine storm that induces, and maintains HLH. In children with predominantly hereditary disease, the HLH-94 protocol can be regarded as a “one size fits all” algorithm. HLH in adults is a much more heterogeneous syndrome requiring a more individualized approach depending on the underlying trigger, disease severity and course, as well as genetic background. Additionally, treatment in adults usually needs to be modified in the face of the preceding disease history and comorbidities. Interdisciplinary patient care with rheumatologists, gastroenterologists, neurologists, pediatricians, the transplant team, and pathologists is a prerequisite to successful treatment. The preferred approach should reflect a disease- and risk-adapted treatment that includes rigorous supportive care with continuous reassessment of sequential therapeutic measures. It should be recognized that the algorithm of HLH treatment in adults is based more on expert opinion than on extensive scientific evidence.
Learning Objectives
Treating HLH is not treating a specific disease, but instead is aimed at treating an overt inflammatory immune response to various triggers
Pediatric treatment algorithms need critical adaptation for adults
Prolonged immunosuppression in aHLH should be avoided
General thoughts
Hemophagocytic lymphohistiocytosis has a broad clinical phenotype overlapping with systemic inflammatory response syndrome (SIRS), sepsis, and multiorgan dysfunction syndrome. Patients are frequently diagnosed with HLH in intensive care units (ICUs), where they are treated for a life-threatening condition classified as “sepsis”. 1 Making the correct diagnosis is, as always, key to optimal treatment, but may be more challenging in the setting of HLH. Treating HLH in adults is not treating a disease solely as a specific entity, but is focused on treating a deranged immune response to various triggers/diseases. This includes classical life support measures in ICU, specific treatment of the trigger, and effective suppression of the cytokine storm and deleterious proliferation of cytotoxic CD8+ T cells and macrophages. Patients may suffer from severe coagulopathy and thrombocytopenia, and bleeding needs to be proactively addressed. Prophylactic antimicrobial treatment and infection control measures for highly immunocompromised patients are pivotal, as treatment usually aggravates the intrinsic disease-associated severe immune-dysfunction of such patients. Initiation of therapy prior to progression of the disease to the point of irreversible organ damage is a major prognostic factor.2 None of the diagnostic criteria for HLH is specific and many patients may express some of the diagnostic features in less than clear cut ways with gradual clinical deterioration leading to a decision to start HLH-treatment based more on strong clinical suspicion, rather than on unequivocal evidence. In many instances, HLH is diagnosed with an immediate need for treatment due to imminent respiratory, hepatic, renal, or hematopoietic failure, without a definitive diagnosis as to whether HLH has a hereditary background (degranulation assay and/or mutation analysis reports pending). Control of overt inflammation is of utmost importance. Classification of HLH as primary (hereditary) or secondary disease has no impact on induction treatment, but rather helps to stratify consolidation therapy. Thus, treatment delay due to outstanding laboratory studies should be avoided.
Nevertheless, one should attempt to correlate HLH to the most likely background condition or trigger in order to choose optimal initial and sequential treatment modalities and to protect the patient from harmful therapies. The largest literature survey on adult HLH from a global perspective found 41.1% of aHLH to be triggered by infections, and 38.8% by malignancies.3 Every effort must be made to detect triggering infections [Epstein Barr virus (EBV), cytomegalovirus (CMV), human immunodeficiency virus (HIV), influenza, bacteria, fungi, and protozoa] that are amenable to treatment.4 In particular Leishmania should not be overlooked, as specific treatment with liposomal amphotericin effectively reverts Leishmania-associated reactive HLH.5 A workup for neoplastic disorders, in particular lymphomas, is mandatory. Does the patient have a history of iatrogenic immunosuppression due to autoimmune disease or due to cancer treatment? Is the malignancy in remission? In a French retrospective multicenter study with 162 patients, about 50% had underlying immunosuppression.6 The heterogeneity of these conditions makes it clear that a “one size fits all” protocol does not make sense for aHLH. It must be emphasized that the pediatric protocols HLH-94 and HLH-2004 have been developed specifically for children, a large percentage of whom suffer from hereditary HLH, need prolonged treatment, and are frequently on track for allogeneic transplant.7 When searching HLH-treatment guidelines for adult patients, one is easily directed to pediatric treatment recommendations, which are not always designated as such; ie, www.uptodate.com/contents/treatment-and-prognosis-of-hemophagocytic-lymphohistiocytosis; accessed April 27, 2015). However, data from prospective trials for adults are not available. In particular, neither HLH-94 nor HLH-2004 have been examined for efficacy or toxicity data in adult patients.
Treatment recommendations for adult HLH patients can thus only be derived from expert opinion The present recommendations are based on a literature survey, retrospective observational studies, and the author's personal experience.2,6,8-16 We have initiated the German adult HLH registry, which offers consultation for physicians treating adult HLH patients (www.hlh-registry.org). As adult HLH is a rare disease, and most available information refers to guidelines for children, contacting the respective national adult HLH reference center is strongly recommended. In Germany, the pediatric HLH-center at the University Hospital Hamburg-Eppendorf (www.uke.de/kliniken/haematologie/index_66512.php) works closely with the adult HLH center at the UK of Jena to aid in diagnostics, and to avoid biased recommendations from an inappropriate perspective (ie, pediatric vs adult medicine). The Histiocyte Society HLH steering committee has launched a focused adult HLH working group aimed at building an international adult HLH registry, to develop recommendations tailored for adult HLH patients, and to foster international networking of the respective study centers (www.histiocytesociety.org).
The roots of HLH-treatment
Epipodophyllotoxins, corticosteroids, and T-cell directed immunomodulation
Prior to use of the epipodophyllotoxins etoposide and teniposide in infant HLH, single cases with prolonged response to weekly vinblastine and corticosteroids (CSs) were reported.17 At that time, HLH was usually fatal with an average survival of two months. In 1980, Ambruso et al from Denver Children's Hospital reported the successful use of etoposide (VP-16) in 2 patients who failed vinblastine.18 Henter et al from the Karolinska Institute (Sweden) published four patients with striking response to teniposide as induction treatment.19 It has recently been demonstrated that etoposide potently and selectively deletes activated T cells, and suppresses inflammatory cytokine production in a murine perforin-deficiency model.20 As pathophysiologic data suggested a predominant T cell-dependent immune-dysregulation, the French pediatric HLH-group focused on T-cell directed immunomodulation using methylprednisolone (2-5 mg/kg initially, with gradual tapering), 10 mg/kg rabbit antithymocyte globulin (rATG) for 5 days, and cyclosporine A (CSA; 150-200 ng/mL trough level) as a maintenance strategy.21 Of note, CS were progressively tapered within 2 weeks, CSA-treatment started on day 16, and 5 doses of intrathecal MTX (6 mg) were administered within 6 weeks to control CNS-involvement. This approach achieved remission even in a patient with chemo-refractory disease, but more often failed when patients were not responsive to VP16/Dex.21 Subsequently the HLH Study Group of the Histiocyte Society22 launched the HLH-94 protocol, which combined etoposide induction with dexamethasone (DEX), and added CSA as maintenance/bridging therapy. This approach allowed extended treatment of patients with secondary HLH and continued response, or provided a bridging therapy to consolidation treatment by allogeneic stem cell transplantation in hereditary or relapsed disease.23 This protocol significantly improved outcome. With a 6.2 year median follow-up, the 5 year survival was 54% in 249 patients <16 years old. Because 29% of patients died prior to transplant, 97% of whom died from active disease, the HLH-2004 protocol aimed to enhance induction treatment by starting CSA simultaneously with etoposide thereby suppressing T-cell proliferation and cytokine secretion, and by adding hydrocortisone to intrathecal treatment.7 This trial is awaiting final analysis. Secondary infections acquired during the course of HLH-treatment seem to be the leading cause of death besides treatment refractory HLH (G. Janka, Pediatric HLH-Registry, personal oral communication, April 29, 2015). It is therefore important to continuously reassess the need for immunosuppressive therapy, and to avoid prolonged immunosuppressive therapy in patients with acquired HLH in remission (see Figure 2). The HLH-equivalent macrophage activation syndrome (MAS) in patients with autoimmune/autoinflammatory diseases (AIDs) needs specific consideration, as MAS-treatment relies on methylprednisolone pulse therapy with CSA or specific treatment of the underlying disease, and rarely requires HLH-specific treatment.24
Intravenous immunoglobulins
The role of intravenous polyvalent immunoglobulins (IVIG) in HLH is twofold: as support for defective humoral immunity in disease or treatment-dependent immune-deficiency (replacement dosing), or as part of the targeted anti-inflammatory treatment regimen (therapeutic dosing). Therapeutic dosing requires high doses (up to 1.6 g/kg over 2-3 days) and acts through diverse mechanisms, ie, Fc-receptor blockade, inhibition of complement activation, or neutralization of cytokines.25,26 There is no consensus on whether IVIG is active in specific subtypes of HLH, or not in others (lymphoma associated, EBV-triggered); whether its efficacy depends on time of application to block macrophage activation (ie, at ferritin peak), or whether the distinct pathophysiology of HLH in children versus adults explains differential efficacy observed in these populations.27 As there is usually a time lag to HLH diagnosis and HLH-specific treatment, and many adult HLH patients develop HLH on the background of immunosuppression, we routinely administer IVIG at 1-1.6 g/kg over 2-3 days in combination with CS (prednisolone 1-3mg/kg KG), and continue replacement therapy in cases with Ig-deficiency. In patients with a transient episode of HLH (ie, infection associated HLH), pulsed treatment with CS/IVIG alone often is sufficient to control HLH.
Stem cell transplantation
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative therapy for hereditary HLH. Fisher et al were the first to report successful sibling allogeneic stem cell transplantation in a 25-month-old child with chemo-refractory familial HLH (FHL).28 Even though the pathophysiology of FHL was not well-defined at that time, this work showed that defective immunoregulation in hereditary HLH can be reversed effectively by HSCT. However, in larger series, myeloablative conditioning (MAC) was associated with a high mortality rate (35% at 100 days).29 It was also demonstrated that remission prior to transplant is a major prognostic factor. When induction treatment fails, alemtuzumab in a median dose of 1 mg/kg split over a median of 4 days may induce remission prior to transplant.30,31 Subsequent trials used reduced intensity conditioning (RIC), mostly containing fludarabine, melphalan or treosulfan, and alemtuzumab, which reduced toxicity and significantly improved 3 year survival from 43% (MAC) to 92% (RIC).32,33 Experimental data from the Cincinnati group using a perforin-deficient mouse model revealed that a 10%-20% perforin expression in animals with mixed chimerism stably re-establishes immune competence, allowing effective negative-feedback and inhibition of cytotoxic cells through killing of dendritic cells.34 This animal model further supports RIC-HSCT as rational approach to HLH.
RIC-HSCT is the standard of care in pediatric centers, and is recommended for children with hereditary disease.35 In the prospective HLH-94 protocol, which contained stem cell therapy for FHL or relapsed HLH in children <16 years old, long-term cure was achieved in more than 50% of patients. In adults, standardized treatment cannot be established for the following reasons: (1) hereditary HLH with known genetic defects is rare (between 7% and 14%),36-38 (2) the clinical course of patients carrying late-onset hypomorphic HLH-mutations is variable and often more benign, and (3) HLH activity in many cases is maintained by an underlying trigger that mandates specific treatment. Consequently, the use of HSCT as consolidation in adults should be decided on a case-by-case basis. Although HSCT has been implemented successfully for aHLH, evidence is limited to case reports or retrospective series.9,37 The largest adult HLH cohort has been collected by the Chinese HLH workgroup, which included 613 patients from 46 hospitals. HSCT was performed in 45/613 (7.3%) cases with a treatment related mortality of 12/45 (26.7%), treatment failure and death in 7/45 (15.6%), and a complete remission rate of 26/45 (57.8%).37 The final publication of these data are forthcoming and should provide more details on the type of transplant and long-term outcome.
The HLH-94 and HLH-2004 protocols in adult HLH
When should the HLH-94 or HLH-2004 protocols be used for treatment in adults? The HLH-2004 protocol, which moved CSA into upfront induction treatment simultaneously with etoposide and DEX should not be administered routinely. Because of increased neurotoxicity,39 and absent the final analysis of HLH-2004, the current recommendation is not to use CSA in the first couple of weeks. Currently, the HLH-94 protocol, perhaps with individual adaptation, may be used in the following: (a) severe aHLH with unknown trigger, (b) known hereditary HLH (with the full picture of severe HLH), and (c) relapsed HLH (relapse has to be defined by strict criteria in order to exclude infectious episodes/sepsis that may mimic HLH).
The term HLH-94-like treatment frequently is applied in situations, where single elements (etoposide, DEX, CSA, IVIG, i.th. MTX) are used on a case-by-case basis, to overcome overt inflammation with imminent respiratory, hepatic, renal, or hematopoietic failure. For example, Henter et al have proposed a modified HLH-94 protocol for patients with reactive HLH infected by the avian flu (H1N5) that contained tapered DEX doses with weekly (×8) etoposide infusions (Figure 1).40 The rationale stems from successful treatment of EBV-associated HLH (EBV-HLH) by the HLH-94 protocol, and the notion that timely administration (<4 weeks from diagnosis) of etoposide is essential to prevent irreversible organ damage by stopping proliferation of cytotoxic CD8+ lymphocytes and macrophages.2,41 The authors emphasize the importance of age adapted dosing (100 mg/m2 down to 50 mg/m2 in elderly patients), as prolonged etoposide-induced bone marrow suppression impairs immune recovery, and increases the risk for secondary infections.

Treatment algorithm for VAHLH (influenza) according to Henter et al.40 Note in red the reduced VP-16 dose as proposed for patients >15 years.
Treatment algorithm for VAHLH (influenza) according to Henter et al.40 Note in red the reduced VP-16 dose as proposed for patients >15 years.
Hepatic impairment with prominent cholestasis is often a matter of concern. Etoposide is metabolized by the liver, however, major excretion is by the kidney42 ; timely application of etoposide reverses histiocytic proliferation and cytokine storm, which cause dysfunctional bile ducts.43 Delay in therapy may carry a risk of marked biliary cirrhosis and liver failure. We administer 50%-75% dose-reduced etoposide and increase the dose as liver function improves. In the setting of renal failure, dose-reduced etoposide [creatinine clearance (CrCl) <50 mL/min 25%; CrCl <15 mL/min 50%) is administered. In our institution, we regard 100 mg/m2 as the maximum etoposide dose for aHLH.
Data on the incidence of CNS-involvement in aHLH are not available. We do not routinely administer intrathecal MTX/CS, but evaluate CNS status by magnetic resonance imaging and lumbar puncture at baseline if the patients present with neurologic symptoms.
The HLH-94 algorithm should not be used for patients with MAS, where pulse methylprednisolone (30 mg/kg for 3 days followed by 2-3mg/kg/day in 2-4 divided doses) is the standard of care.24 CSA is recommended in the event of insufficient response, alternatively tacrolimus may be selected.44 In patients with MAS failing this treatment, the interleukin-1 receptor antibody Anakinra has been used successfully.45 All patients with MAS should be evaluated for infectious triggers.
Virus associated HLH (VAHLH)
EBV is the most frequent trigger of childhood FHL, with the largest series reported in Asia.46 The prognosis was excellent when promptly treated by HLH-94. When aHLH is associated with a viral infection, treatment differs between patients with pre-existing immunosuppression and apparently immunocompetent patients. One should prescreen for the presence of a genetic defect by degranulation and expression assays with subsequent genotyping as recommended.35 In the setting of late onset hereditary HLH, HSCT needs strong consideration. However, depending on the severity and course of HLH, a more conservative approach with short course CS/IVIG is justified in patients with less severe disease.
As EBV replicates predominantly in B cells, treatment with CS and rituximab is often effective.47 When treatment beyond CS/IVIG is necessary, we add rituximab to HLH-specific treatment components at 375 mg/m2 once weekly (2-4×). As T-cell infection by EBV is often seen in EBV-related HLH, monitoring of EBV viral load, ferritin, and sCD25 is required to detect failing patients.48 The wide spectrum of potential viral triggers mandates virus-specific treatment on a case- by-case decision.
HIV and aHLH
The prognosis of aHLH in HIV-patients has improved in the HAART era.49 As HIV-patients are predisposed to the development of lymphoma and opportunistic infections, these are the most important triggers that should be sought. Specific treatment with focus on drug–drug interactions, and short transient treatment of overt inflammation by CS/IVIG is recommended. aHLH has also been observed in the context of immune reconstitution syndrome after introduction of HAART.
Treatment of malignancy-associated HLH (MAHLH)
Malignant disease with MAHLH has the worst prognosis of all aHLH subgroups.10,37 The risk of developing MAHLH increases with age.50 Lymphoma associated HLH (LAHLH) is the major cause for MHLH with region-specific variation in subtype distribution (eg, with an increased rate of NK/T-cell and EBV-triggered lymphoma in Asia). Treatment of MAHLH needs to balance HLH-specific and tumor-specific treatment. CSs are used to combat inflammation. IVIG may be added. If severe organ damage by inflammatory lymphoproliferation is imminent, dose adjusted etoposide (50-100 mg/m2) may be used to control HLH prior to start of tumor-specific treatment. It is reasonable to add etoposide to the CHOP-protocol (CHOEP) for lymphoma treatment. In eligible patients in remission, high-dose consolidation chemotherapy with autologous stem cell transplantation (ASCT) is standard of care. A decision for allogeneic transplantation requires careful individual assessment.51 As the risk of lymphoma is increased in patients with certain hereditary defects of degranulation (perforin/PRF1, XLP1/SAP, Munc 18.2/STXBP2),50 coincidence of HLH and lymphoma, particularly EBV-driven lymphoma, should trigger expert consultation and guided genotyping, as the finding of disease causing FHL-mutations further supports a decision toward HSCT.52 HLH in an adult patient with a malignancy represents an additional adverse prognostic factor that needs to be accounted for in choosing consolidation treatment. We and others therefore recommend early HLA-typing and donor search in MAHLH.44
Chemotherapy associated aHLH
It is important to discriminate between a malignant disease presenting with concomitant HLH, and HLH that develops in the course of treatment for a malignant disorder. In neutropenic patients after induction chemotherapy for AML, as many as 10% may develop aHLH with infections as the most frequent trigger.13 HLH complicating cancer treatment is probably under-recognized. Diagnosis is obscured by pre-existing neutropenia; liver functional abnormalities may be attributed to toxic drug effects; ferritin elevation may also be attributed to transfusion-related iron-overload; and neutropenic fever is the most prevalent complication of induction chemotherapy. However, HLH patients suffer from prolonged neutropenia, pulmonary, neurologic symptoms, liver abnormalities, and lower platelet counts, and need anti-inflammatory treatment with CS (1-2 mg/kg KG) ±IVIG (1.6 g/kg KG over 2-3 days), and adapted antimicrobial treatment to treat the infectious trigger. Etoposide should be used sparingly, as bone marrow recovery is central for immune reconstitution. Ongoing monitoring is required to detect recurrent malignant disease as a potential alternate trigger for HLH. I have found laboratory tests for sCD25 and bone marrow assessment for hemophagocytosis to be helpful for this challenging differential diagnosis.
Treatment of HLH after HSCT needs to be distinguished from engraftment syndrome and graft-versus-host-disease, and frequently coincides with viral reactivation.53 CS, IVIG, and specific antibiotic treatment are warranted.
Monitoring and supportive therapy
Impaired biliary excretion, renal failure, respiratory distress, and severe bleeding due to hypofibrinogenemia, disseminated intravascular coagulation, and thrombocytopenia need immediate action in a patient presenting with HLH. Blood products and coagulation factors are supplemented as needed. Patients deteriorating while on HLH-treatment need to be evaluated for secondary infections. Measuring C-reactive protein (CRP) may be helpful, as it may serve as marker for secondary infections. We perform weekly monitoring for opportunistic infections (galactomannan, CMV-PCR) und provide antimicrobial prophylaxis against pneumocystis and fungi. Stringent clinical assessment for infections with regular examinations of patients is mandatory. Because G-CSF has been reported in case reports to reactivate inflammation, and can induce capillary leak syndrome, we do not prescribe growth factors in active HLH, but use them in patients with low inflammatory activity under chemotherapy for MAHLH. See Figure 2 for the proposed aHLH treatment algorithm.

Treatment algorithm for aHLH. Focus of aHLH-mangement is detecting and treating the trigger disease. HLH-specific treatment (VP-16, CSA, IVIG) is required in severe HLH prior to prevent irreversible organ damage (liver, kidney, or respiratory failure). Hourly/daily reassessment is mandatory. Close contact to HLH-reference centre is recommended; *Indicates treatment with CAR T cells or T-cell engaging antibodies; §, off label; αCD20, Rituximab; αIL1R, Anakinra; αIL6R, Tocilizumab; MPPT, methylprednisolone pulsed therapy; Pt, patient; and VP-16, etoposide.
Treatment algorithm for aHLH. Focus of aHLH-mangement is detecting and treating the trigger disease. HLH-specific treatment (VP-16, CSA, IVIG) is required in severe HLH prior to prevent irreversible organ damage (liver, kidney, or respiratory failure). Hourly/daily reassessment is mandatory. Close contact to HLH-reference centre is recommended; *Indicates treatment with CAR T cells or T-cell engaging antibodies; §, off label; αCD20, Rituximab; αIL1R, Anakinra; αIL6R, Tocilizumab; MPPT, methylprednisolone pulsed therapy; Pt, patient; and VP-16, etoposide.
Future directions
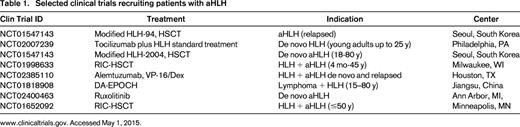
Current clinical trials are evaluating the addition of rATG (NCT01104025), the anti-interleukin 6 antibody tocilizumab (NCT02007239), or the anti-CD52 antibody Alemtuzumab to induction treatment with etoposide/dexamethasone (NCT02385110). These approaches are intended to enhance treatment activity against the inflammatory cytokine storm without further impairing hematopoietic recovery. Innovative approaches addressing molecular inhibition of the cytokine signaling mediator Janus-kinase1/2 (JAK1/2) by Ruxolitinib (NCT02400463), or targeting the inflammatory cytokine interferon-gamma by monoclonal antibody NI-0501 (NCT02069899) are in early clinical development (Table 1).
As we become more vigilant, we will encounter more patients with aHLH. As more immunomodulatory drugs and treatment modalities become available, and cancer treatment achieves more long-term remissions with chronic immunomodulatory treatment of the disease, we are likely to see a rise in aHLH incidence in immunosuppressed patients. Examples are reports on secondary HLH after treatment with blinatumomab or after treatment with chimeric antigen receptor (CAR) T cells.54,55 In this situation immediate treatment with the IL-6 antibody tocilizumab can overcome severe cytokine release, leading to HLH. As in the case with CAR T cell, where CS may interfere with therapeutic efficacy (proliferating anti-cancer T cells), specific expertise in the treatment of HLH becomes important as therapeutic options evolve. Biologic treatment needs systematic evaluation, and clinical trials aiming at improving HLH-treatment for adults are recruiting (Table 1). HLH is an orphan disease where close interaction with the national study centers is highly recommended. The registry initiative of the Histiocyte Society has the goal of collecting data on aHLH, which will allow the establishment of formal treatment guidelines derived from evidence and consensus.
Acknowledgments
The author thanks G. Janka, Dr K. Lehmberg, and S. Ehl from the German pediatric HLH-center for supporting the aHLH-initiative, and continuous advice, and fruitful discussions, and G. Janka in particular for critically discussing the paper.
This work was supported by Europa für Thüringen – Europäischer Sozialfonds 2014 KN 0024, and is part of the campaign agenda of the working group adult HLH of the Histiocyte Society.
Correspondence
Paul La Rosée, Klinik für Innere Medizin II. Abt. Hämatologie u. internistische Onkologie, Erlanger Allee 101, 07747 Jena, Germany; Phone: +49-3641-9-324201; Fax: +49-3641-9-324202; e-mail: paul.larosee@med.uni-jena.de.