Abstract
Large granular lymphocyte (LGL) leukemia represents a spectrum of rare lymphoproliferative diseases defined by clonal amplification of either CD3+ cytotoxic T-lymphocytes or CD3− natural killer cells. This chapter focuses on the T-cell form of LGL leukemia. Clinical features include neutropenia, anemia, and rheumatoid arthritis. LGL leukemia is thought to arise from chronic antigenic stimulation, with the long-term survival of LGL being promoted by constitutive activation of multiple survival signaling pathways, such as the JAK/STAT3, sphingolipid, and Ras/MEK/ERK pathways. Therefore, these lead to global deregulation of apoptosis and resistance to normal pathways of activation-induced cell death. The majority of LGL leukemia patients eventually need treatment. Treatment of leukemic LGL is based on immunosuppressive therapy, primarily using low doses of methotrexate or cyclophosphamide. However, no standard therapy has been established because of the lack of large, prospective trials. In addition, because some patients are refractory to currently available treatments and none of these therapeutic modalities can cure LGL leukemia, new therapeutic options are needed. Understanding the current state of the pathogenesis of LGL leukemia may provide insights into novel therapeutic options.
Introduction
Large granular lymphocytes (LGLs) are large WBCs (15-18 μm) with reniform or round nuclei and abundant cytoplasm that contains typical azurophilic granules. In normal adults, LGLs account for 10%-15% of peripheral blood mononuclear cells (PBMCs). This population of cells can be further classified into 2 different lineages of lymphocytes based on their cell surface markers: CD3− natural killer (NK) cells and CD3+ cytotoxic T lymphocytes (CTLs). LGL leukemia represents a rare chronic lymphoproliferative disorder of CTLs, a malignancy that involves lymphocyte infiltration of multiple organs, including the BM, liver, and spleen. Phenotypically, LGL leukemia can arise from either CD3+ CTLs or CD3− NK cells.1 The World Health Organization (WHO) classification includes T-cell LGL leukemia in the subgroup of mature peripheral T-cell neoplasms and distinguishes it from aggressive NK-cell leukemia. In addition, a new provisional entity of chronic lymphoproliferative disorder of NK cells was created by the WHO in 2008. Many features of molecular pathogenesis and clinical presentation of T-LGL leukemia are similar to those of chronic lymphoproliferative disorders of NK cells.
Clinical manifestations and diagnosis
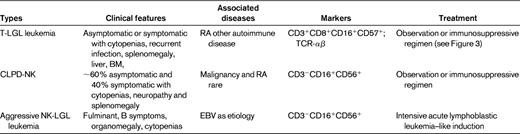
This chapter focuses on T-LGL leukemia (referred to hereafter as LGL leukemia). Table 1 summarizes clinicopathologic features of this disease, as well as the NK disorders that should be considered in the differential diagnosis. LGL leukemia occurs equally in men and women with typical age of onset of approximately 60 years. Approximately one-third of patients are asymptomatic at diagnosis, whereas two-thirds of patients will become symptomatic during the course of their disease. Predominant symptoms include neutropenia, anemia, and rheumatoid arthritis (RA).2 Eighty-five percent of patients with LGL leukemia experience neutropenia, and 45% develop severe neutropenia (absolute neutrophil count [ANC] < 500/μL).3 Recurrent bacterial infections are a hallmark of LGL leukemia as a consequence of neutropenia. Anemia is becoming more frequently recognized and transfusion dependence may occur in 5%-35% of patients.3 There appear to be multiple mechanisms of anemia, including autoimmune hemolysis and pure RBC aplasia. RA has been reported in every series of LGL leukemia patients,2 and is usually diagnosed before the onset of LGL leukemia. Activating STAT3 mutation is associated preferentially with the LGL/Felty syndrome phenotype (ie, neutropenia and RA).4 Splenomegaly is seen in 20%-50% of patients; thrombocytopenia is less common, being detected in 20% of patients.3
Patients with unexplained cytopenias or RA should be tested for circulating LGLs. Diagnosis of LGL leukemia is established by documentation of an increased number of clonal LGLs (Figure 1).5–7 LGL counts can be determined by peripheral blood smear evaluation and/or flow cytometry. The normal number of LGLs in the peripheral blood is < 0.3 × 109/L. Initially, a circulating LGL count > 2 × 109/L was considered as mandatory,1 but now a lower count (range, 0.4-2 × 109/L) can be compatible with the diagnosis in the proper clinical setting. Phenotypic analyses have revealed that the leukemic LGL cells are terminal effector memory T cells (CD45RA+CD62L−).8 The majority (80%-90%) of patients with T-LGL leukemia show a CD3+CD8+CD57+CD56−CD28−, TCR-αβ+ phenotype.3 Uncommon variants include CD4+ with or without coexpression of CD8. Less than 10% of patients have TCR-γΔ, which is clinically similar to TCR-αβ, and have a favorable survival of 85% at 3 years.9 Clonality is readily ascertained by detecting TCR gene rearrangement using Southern blotting and/or PCR. More recently, flow cytometry has been used to assess clonality by demonstrating a predominant expression of a TCR Vβ family, because mAbs now are available for approximately 70% of the variable region families of the TCRβ chain.10 In patients with low LGL counts, we recommend BM aspirate and biopsy to aid in diagnosis. Pathologic findings of lymphoid interstitial infiltration with linear arrays of CD8+, TIA-1 (+), and granzyme B (+) supports the diagnosis of LGL leukemia.

How to establish the diagnosis of LGL leukemia. The diagnosis is based on a LGL peripheral expansion (> 0.5 × 109/L). Specific criteria for T-LGL leukemia include expression of LGL surface markers compatible with an activated T-cell (commonly CD3+/CD8+/CD57+ and/or CD16+) phenotype and clonal rearrangement of the TCR-γ gene using PCR or specific and clonal Vβ expression using flow cytometry. Specific criteria for NK-LGL leukemia and NK-LGL lymphocytosis include expression of LGL surface markers compatible with an NK-cell (commonly CD3−/CD8+/CD16+ and/or CD16+/CD56+) phenotype. The term chronic NK-LGL lymphocytosis is used for patients with relatively few symptoms and chronic illness, whereas patients with massive tissue LGL infiltration of the spleen, liver, and BM and presenting aggressive clinical behavior are considered to have NK-LGL leukemia. Reproduced with permission from Lamy and Loughran.6
How to establish the diagnosis of LGL leukemia. The diagnosis is based on a LGL peripheral expansion (> 0.5 × 109/L). Specific criteria for T-LGL leukemia include expression of LGL surface markers compatible with an activated T-cell (commonly CD3+/CD8+/CD57+ and/or CD16+) phenotype and clonal rearrangement of the TCR-γ gene using PCR or specific and clonal Vβ expression using flow cytometry. Specific criteria for NK-LGL leukemia and NK-LGL lymphocytosis include expression of LGL surface markers compatible with an NK-cell (commonly CD3−/CD8+/CD16+ and/or CD16+/CD56+) phenotype. The term chronic NK-LGL lymphocytosis is used for patients with relatively few symptoms and chronic illness, whereas patients with massive tissue LGL infiltration of the spleen, liver, and BM and presenting aggressive clinical behavior are considered to have NK-LGL leukemia. Reproduced with permission from Lamy and Loughran.6
Molecular pathogenesis of LGL leukemia
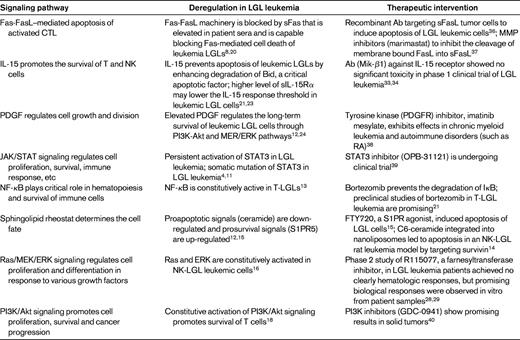
During infection exposure or Ag stimulation, LGLs undergo vigorous proliferation by approximately 50 000-fold upon priming by target cells, and at a later time after Ag clearance, are selectively eliminated by a process called activation induced cell death (AICD). However, in LGL leukemia patients, the AICD process is dysfunctional and activated CTL cells do not undergo apoptosis efficiently, leading to elevated number of LGLs in the peripheral blood. Multiple cell survival pathways, including JAK2/STAT3,4,11 sphingolipid signaling,12–15 RAS/MEK/ERK,16 and SFK/PI3K/Akt,17,18 have been found to be constitutively activated in LGL leukemia patients. A systems biology approach identified IL-15 and PDGF as master survival signaling switches that may have a profound effect on all known deregulations in T-LGL leukemia.13 Most recently, STAT3 mutations were found to be highly associated with LGL leukemia, suggesting that aberrant STAT3 signaling underlines the pathogenesis of LGL leukemia.4 Global deregulation of cell proliferation and survival signaling pathways in LGL leukemia are summarized in Figure 2.

The signaling network underlying LGL leukemia pathogenesis. Up-regulated or constitutively active nodes are highlighted in pink; down-regulated or inhibited signals are in green; the states of white nodes are unknown or unchanged compared with normal. ASAH indicates acid ceramidase; RTK, receptor tyrosine kinase.
The signaling network underlying LGL leukemia pathogenesis. Up-regulated or constitutively active nodes are highlighted in pink; down-regulated or inhibited signals are in green; the states of white nodes are unknown or unchanged compared with normal. ASAH indicates acid ceramidase; RTK, receptor tyrosine kinase.
Deregulation of Fas-FasL–mediated apoptosis in LGL leukemia
Fas, a member of the TNF receptor family, is involved in transducing death signals. The interaction of Fas ligand (FasL) with its receptor (Fas) results in the formation of death inducing signaling complex (DISC) and the activation of many apoptotic effectors. Under physiological conditions, after Ag clearance, activated CTLs are eliminated through the process of AICD in part by the Fas-FasL pathway. However, leukemic LGLs are resistant to this death pathway despite high levels of surface expression of Fas, abundant constitutive expression of FasL, and absence of mutations in the Fas receptor gene.19 In addition, cells from T-LGL leukemia patients exhibit impaired Fas-induced DISC formation after cross-linking of the Fas receptor.8 Further study found that the DISC inhibitory protein cellular Fas-associated protein with death domain (FADD)–like IL-1 converting enzyme-inhibitory protein (c-FLIP) is overexpressed in leukemic LGLs, resulting in reduced DISC assembly and resistance to Fas-FasL–mediated apoptosis.8 Moreover, a soluble form of Fas (sFas) that is capable of blocking Fas-dependent apoptosis was detected in the sera of LGL leukemia patients but not in normal serum.20 It is believed that sFas competes with Fas by acting as soluble decoy receptor, leading to resistance to Fas-mediated apoptosis in leukemic LGLs. Impaired Fas-FasL–mediated apoptosis contributes to the pathogenesis of LGL leukemia.
IL-15
IL-15 is a member of the IL-2 family that regulates T- and NK-cell activation and proliferation by altering expression of Bcl-1 family members (eg, Bcl-2 and Bcl-XL). IL-15 is also required to provide long-term survival signals to maintain normal NK and CD8+ memory T cells13 and plays an important role in the survival of leukemic LGL cells. Inhibition of IL-15 led to apoptosis in leukemic LGL cells,21 whereas interruption of IL-15 signaling using an IL-15–neutralizing Ab was able to prevent LGL leukemia in IL-15–transgenic mice.22 In NK cells, IL-15 specifically reduces the level of BH3 interacting domain death agonist (Bid) by up-regulating the E3 ligase E3 ubiquitin-protein ligase double minute 2 protein (HDM2) that targets Bid to proteasome for degradation.21 Interestingly, low levels of Bid could be reversed with bortezomib (a proteasome inhibitor) treatment, leading to apoptosis of leukemic LGLs.21 Recently, the IL-15 private soluble receptor subunit IL-15Rα has been shown to be up-regulated in the PBMCs of some T-LGL leukemia patients.23 Moreover, PBMCs from some T-LGL patients proliferated at higher levels in response to exogenously added IL-15 compared with normal PBMCs. It has been suggested that the higher level expression of IL-15Rα contributes to the pathogenesis of T-LGL leukemia by lowering the IL-15 response threshold in T-LGL leukemia.
PDGF
PDGF is a major growth factor that has been found to mediate the survival of leukemia LGL of both T- and NK-cell origin. Network modeling predicted a key role for IL-15 and PDGF in LGL leukemia pathogenesis.13 We demonstrated that leukemic LGL survival was dependent on a PDGF autocrine loop. PDGF regulated the long-term survival of leukemic LGLs through the PI3K-Akt and MER/ERK pathways.24
Constitutive activation of STAT3 in LGL leukemia
The JAK/STAT signaling pathway controls a variety of biological processes such as cell proliferation, apoptosis, angiogenesis, and immune responses. STAT3, a latent transcription factor, has been shown to play a central role in conferring cell survival. Persistent activation of STAT3 has been observed in more than 22 types of cancers. STATs can be activated by cytokine and growth factors via engagement with their respective receptors (ie, cytokine receptors and growth factor receptors), leading to dimerization and rapid translocation to nucleus, where they exert their transcriptional activities. In 2001, constitutive activation of STAT3 was found in all cells of LGL leukemic patients (N = 19). STAT3 promoted the survival of leukemic LGL by regulating the expression of antiapoptotic proteins such as myeloid cell leukemia sequence 1 (Mcl-1).11 Recently, genomic studies demonstrated that the reason for constitutive STAT3 activation in 40% (31 of 77) of LGL leukemia patients was activating somatic mutations in STAT3 Src homology 2 domain, a critical region that mediates STAT3 protein dimerization and activation.4 Downstream target genes of STAT3 pathway (eg, IFNGR2, JAK2, and Bcl2L1) were up-regulated in patients with LGL leukemia.4 It is conceivable that STAT3 inhibitors might be ideal targeted therapies for LGL leukemia.
Constitutive activation of NF-κB
NF-κB, a transcription factor, plays a critical role in the hematopoiesis, proliferation, and survival of immune cells. In its inactivated state, NF-κB stays in the cytoplasm with its inhibitor, IκB. Once the NF-κB pathway is activated, IκB is phosphorylated by IκB-kinase complex, leading to the degradation of IκB and the activation of NF-κB. NF-κB has been shown to promote the expression of prosurvival Bcl-2 family member and inhibitor of apoptosis proteins. Interestingly, leukemic LGLs expressed high levels of c-Rel, a member of the NF-κB family,13 and exhibited higher NF-κB activity than normal PBMCs. Pharmacological inhibition of Akt and NF-κB demonstrated that NF-κB acts downstream of the PI3K-Akt pathway to prevent apoptosis through Mcl-1 independently of STAT3.13
Imbalance of sphingolipid rheostat in LGL leukemia
Sphingolipids and their metabolites play important but opposite roles in diverse biological processes such as proliferation, apoptosis, and cell migration. The balance between the proapoptotic (ie, ceramide and sphingosine) and prosurvival (ie, sphingosine-1-phosphate [S1P]) sphingolipids, rather than the absolute amount of these molecules, determines the cell fate (sphingolipid rheostat). Molecular profiling analysis revealed that the sphingolipid rheostat is deregulated in a way that tilts the balance away from proapoptotic molecules (eg, ceramide) and in favor of survival molecules (eg, S1P) in LGL leukemia.12 For example, acid ceramidase (ASAH1) was found to be constitutively up-regulated in LGL leukemia, leading to decreased levels of ceramide and survival of LGL cells.12 Inhibition of ASAH1 or delivery of C6-ceramide into a rat model of NK-LGL led to cell death of leukemic LGLs both in vitro and in vivo.14 In addition, sphingosine kinase 1 (SphK1), a kinase that converts sphingosine to S1P, is overexpressed in LGL leukemia patients. Pharmacological inhibition of SphK1 with SKI-I or S1P using FTY720 induced significant apoptosis in leukemic LGLs.15 Further, the expression of S1P receptors (S1PRs) are elevated. In particular, S1PR5 is found to be constitutively overexpressed in leukemic LGL.12 The deregulated sphingolipid rheostat contributes to the development of LGL leukemia; therefore, molecules targeting the sphingolipid pathway might prove to be successful therapies in LGL leukemia.
Constitutive activation of Ras/Raf/MEK/ERK signaling
The Ras/Raf/MEK/ERK pathway controls many fundamental cellular processes, including cell proliferation, survival, differentiation, apoptosis, motility, and metabolism. The Ras/Raf/MEK/ERK pathway transfers various signals from growth factors (eg, PDGF), G-protein coupled receptors (eg, S1PR5), and NF-κB to either activate transcription factors (eg, Fos and Jun of Ets family) or promotes the transcription of FLIP and Mcl-1 directly. Mutations in critical components (eg, Ras and Raf) of this pathway occur in approximately 30% of all human cancers.
Ras/Raf/MEK/ERK signaling has been reported to be constitutively activated in NK LGL leukemia patients. Inhibition of Ras resulted in the inhibition of ERK and apoptosis of patient cells. Further, inhibition of ERK by inhibitors (eg, PD98059 and U0126) or a dominant-negative form of MEK reduced the survival of patient cells.16 Therefore, these findings demonstrated that the constitutively active Ras/Raf/MEK/ERK pathway contributes to the survival of LGL cells.
Deregulation of the PI3K/Akt pathway
The PI3K/Akt signaling pathway plays a pivotal role in promoting cell proliferation and survival, and therefore cancer progression. PI3K/Akt signaling is activated by growth factors through either Src family kinase (SFK) or the Ras signaling cascade. The main targets downstream of PI3K/Akt include the prosurvival transcription factors NF-κB, Bcl-2 antagonist of cell death, and pro-caspase 9. In LGL leukemia, SFK maintains PI3K in its constitutively active form, which results in the increased phosphorylation of Akt and glycogen synthase kinase-3 (GSK3).18 After activation, PI3K/Akt signaling enhances the survival of T cells by inhibition of Fas clustering and DISC formation. Inhibition of the PI3K/Akt pathway alone leads to spontaneous apoptosis of T-LGL.18 These results suggest that PI3K/Akt pathway activation protects leukemic LGLs from undergoing homeostatic apoptosis.
In summary, various signaling pathways, including the Fas/FasL, IL-15, PDGF, JAK/STAT3, NF-κB, sphingolipid, and Ras/MEK/ERK, PI3K/Akt pathways, are deregulated in LGL leukemia. A variety of therapeutics targeting these deregulated signaling pathways are under clinical study for the treatment of different cancers, raising the possibility of future clinical trials in LGL leukemia (Table 2).
Therapeutic options for LGL leukemia
Currently, there is no standard treatment for patients with LGL leukemia. For asymptomatic LGL leukemia patients with an indolent course, one can consider a wait-and-see approach with a single G-CSF injection to test the potential myeloid progenitor mobilization, which then could be administered urgently at time of neutropenic fever if the patient is G-CSF responsive. For symptomatic patients, indications for immunosuppressive treatment include severe or moderate anemia (ie, transfusion dependent or symptomatic anemia) and severe or moderate neutropenia (ie, an ANC < 500 or recurrent infection associated with higher ANC > 500). Finally, symptomatic patients with RA warrant therapy.7
The therapeutic approach is summarized in Figure 3.7 The mainstay of treatment is immunosuppressive therapy, with single-agent methotrexate (MTX), oral cyclophosphamide, or cyclosporine A reported to have activity in LGL leukemia. We recommend low-dose MTX initially for LGL leukemia patients with neutropenia and/or RA. Currently, MTX (10 mg/m2/wk orally) or cyclophosphamide (100 mg/d orally) is our first choice for LGL patients with anemia. Response to treatment is evaluated by blood count 4 months after the beginning of the treatment. Hematologic complete response (CR) is considered achieved when blood counts return to normal ranges (eg, platelets > 150 × 109/L, ANC > 500/μL, hemoglobin > 12 g/dL, and lymphocytes < 4 × 109/L) and circulating LGL in the normal range. In addition, complete molecular remission is considered achieved when T-cell clone is nondetectable using PCR. Partial response (PR) is defined as an improvement in blood counts in the absence of CR. Treatment failure is defined as failure to achieve either CR or PR within 4 months after starting therapy. If at least a PR is not attained after 4 months of treatment, then therapy is changed to the other agent or cyclosporine (2.5-5 mg/kg twice daily). The following section briefly reviews the available treatments in LGL leukemia.

Algorithm of treatment of LGL leukemia. Adapted with permission from Figure 5 in Lamy and Loughran.6
Algorithm of treatment of LGL leukemia. Adapted with permission from Figure 5 in Lamy and Loughran.6
MTX has been widely used in RA and was first reported to have efficacy of 50% in hematologic CR in the treatment of a small series of LGL leukemia in 1994.25 Other series have demonstrated a response rate of 39%-67%.7 Our recent large, prospective Eastern Cooperative Oncology Group (ECOG) trial showed 39% response rate to MTX as initial therapy.26 In general, MTX is well tolerated. Side effects associated with low-dose MTX include hepatic injury and, rarely, lung dysfunction. MTX is temporarily held or dose adjusted for transaminase values increased above 2-fold. Responding patients are continued on MTX indefinitely.
Cyclophosphamide, an alkylating agent, is a prodrug used to treat various types of cancers and some autoimmune disorders. In French series of LGL leukemia patients, cyclophosphamide showed a favorable overall response rate compared with MTX, at 66% and 55%, respectively.6 These data suggest that cyclophosphamide could be also considered as a first-line therapy.6 It takes up to 1-4 months to achieve any response, so the same 4-month time frame is used to assess response. In patients achieving at least a PR, cyclophosphamide is continued for a total of 9-12 months to avoid rare complications of myelodysplastic syndrome/acute myeloid leukemia, which appears to be dose dependent.
Cyclosporine is another alternative for first-line therapy. We prefer MTX or cyclophosphamide because of the more extensive toxicity profile of cyclosporine.7 Renal function and blood pressure need to be monitored and treatment may be discontinued due to side effects. It is clear that cyclosporine must be continued indefinitely because relapse happens almost invariably just after stopping treatment. Treatment experience with purine analogs is limited to less than 40 reported patients, but the overall response rate is impressive (79% or 30 of 38 patients).27 Considering the advantages of a short period of treatment (from 1 to 4-6 monthly courses), high response rate, mild toxicity, and the potential of inducing durable remission, a clinical trial using purine analogs could be considered.
There have been a limited number of small clinical trials for LGL leukemia. R115077 (tipifarnib; trade name Zarnestra) is a farnesyltransferase inhibitor designed to inhibit Ras-mediated signaling. A phase 2 clinical study of 8 LGL leukemia patients achieved no clinical hematologic responses.28 However, promising biological responses, including decreased LGL in the blood and BM, improved BM hematopoiesis, and increased hematopoietic colony growth in vitro were observed in most of these patients. In addition, R1150777 treatment improved the pulmonary hypertension symptoms in a LGL leukemia patient.29
Alemtuzumab, the humanized anti-CD52 mAb, is capable of selectively killing CD52-expressing cells. Interestingly, CD52 is expressed on the leukemic T-LGLs. In one study, the overall response rate of alemtuzumab was 50% (4 of 8 T-LGL leukemic patients).30 Currently, alemtuzumab is being studied in a phase 2 clinical trial for treatment of T-LGL leukemia.31 It was approved by US Food and Drug Administration (FDA) as second-line therapy for CLL patients who have been treated with alkylating agents and who have failed fludarabine therapy.32
Humanized MiK-β1 mAb is directed toward CD122, a common subunit of IL-2R and IL-15R. Recently, a phase 1 clinical study has been completed for humanized MiK-β1 mAb in patients with T-LGL leukemia.33 Although down-regulation of the IL-15β subunit was observed in leukemic LGL, no reduction in LGL counts were observed.34
Recently, the efficacy of extracorporeal photopheresis was evaluated in 5 refractory/relapsed patients with LGL leukemia, with 2 of 5 patients achieving a CR.35 Because limited options are available for these refractory/relapsed patients, extracorporeal photopheresis should be considered. Other drugs targeting the signaling pathways that are deregulated in LGL leukemia are listed in Table 2. Such therapeutics could be considered as candidates for clinical trials.
Summary
LGL leukemia is a clonal disorder of CTLs. The mainstay of treatment for LGL leukemia is immunosuppression; however, there are no curative therapeutic modalities for this disease and new therapeutic options are needed. Recognition of many important deregulated signaling pathways has greatly stimulated the identification of potential new therapeutic targets in LGL leukemia. It is hoped that clinical trials using new drugs targeting the deregulated signaling pathways will soon be available for patients with LGL leukemia.
Disclosure
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: immunosuppressive therapies for LGL leukemia that could include MTX, cyclophosphamide, and cyclosporine.
Correspondence
Thomas P. Loughran Jr, Penn State Hershey Cancer Institute, Rm 4427, 500 University Dr, PO Box 850, Hershey, PA 17033-0850; Phone: 717-531-4034; Fax:717-531-0490; e-mail: tloughran@hmc.psu.edu.