Abstract
Neutropenia is defined as the reduction in the absolute number of neutrophils in the blood circulation. Acute neutropenia is a relatively frequent finding, whereas disorders of production of neutrophils are quite rare. Acute neutropenia is often well tolerated and normalizes rapidly. Neutropenia arising as a result of underlying hematologic disorders is far more significant. Such a patient may be at risk for infectious complications and will likely require a thorough investigation. Acute neutropenia evolves over a few days and occurs when neutrophil use is rapid and production is impaired. Chronic neutropenia may last for 3 months or longer and is a result of reduced production, increased destruction, or excessive splenic sequestration of neutrophils. Neutropenia may be classified by whether it arises secondarily to causes extrinsic to BM myeloid cells, which is common; as an acquired disorder of myeloid progenitor cells, which is less frequent; or as an intrinsic defect arising from impaired proliferation and maturation of myeloid progenitor cells in the BM, which is rare. Severe neutropenia with absolute neutrophil counts below 500/μL increases susceptibility to bacterial or fungal infections. Multiple disorders of severe congenital neutropenia have been found by the discovery of genetic defects affecting differentiation, adhesion, and apoptosis of neutrophil precursors. Elucidation of the multiple genetic defects have provided insight into the biology of the cell involving membrane structures, secretory vesicles, mitochondrial metabolism, ribosome biogenesis, transcriptional regulation, and cytoskeletal dynamics, as well as the risk for myelodysplasia and acute myeloid leukemia.
Introduction
Neutropenia is a reduction in the absolute number of neutrophils (segmented cells and bands) in the blood circulation. Normal values for the total WBC and absolute neutrophil count (ANC) change from childhood into adolescence. Values of the ANC from 1 year of age slowly increase throughout childhood until the adult value is achieved during adolescence. Normal neutrophil counts must be stratified for age and ethnicity. The lower limit of the ANC is 1000/μL in white children 2-12 months of age and 1500/μL at more than 12 months of age. Individuals of African descent and some Middle-Eastern ethnic groups may have lower neutrophil counts. The leukopenia and relative neutropenia does not predispose these individuals to infection. Genetic studies in individuals of African descent have been linked to a polymorphism in the gene encoding the Duffy-Ag receptor for chemokines (DARC).1 The Duffy-null polymorphism is associated with protection against invasion of RBCs by the Plasmodium vivax malaria. The absence of the DARC Ag prevents the subsequent invasion of the parasite into Duffy-negative RBCs. Although the mechanism for the association of neutropenia with a lack of DARC on RBCs is not known, it is possible that DARC expression regulates neutrophil storage within the BM via the release of cytokines and chemokines.
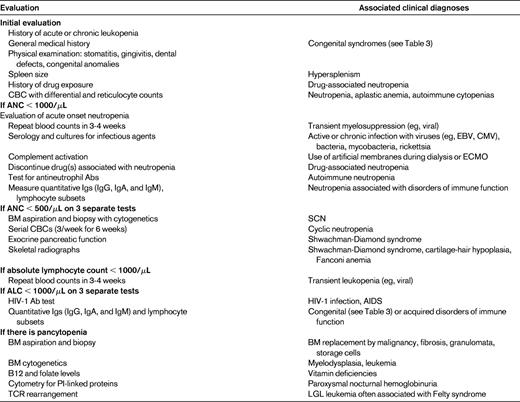
Neutropenia may be characterized clinically as mild neutropenia with an ANC of 1000-1500/μL, moderate neutropenia with an ANC of 500-1000/μL, or severe neutropenia with an ANC of less than 500/μL.2 This stratification aids in predicting the risk of pyogenic infections in patients with chronic neutropenia; only patients with severe neutropenia are at risk for major pyogenic infections and life-threatening infections. Severe neutropenia is chronic if it lasts more than 3 months and places the patient at risk for pyogenic infection.2 Normally, the neutrophil count fluctuates physiologically in a nonrandom fashion and is subject to variation; therefore, neutropenia should ideally be confirmed on at least 3 samples obtained over several weeks.3 Evaluation of patients with neutropenia begins with a thorough history, physical examination, family history, and screening laboratory tests (Table 1). BM aspiration is indicated in patients with severe chronic neutropenia, pancytopenia, or severe infection.4
Epidemiology
Neutropenia is a relatively frequent finding, whereas congenital and cyclic neutropenia are quite rare. All forms of congenital neutropenia, including cyclic neutropenia, occur at 6.2 cases per million according to the French National Registry of Primary Immunodeficiency Diseases.3 Both congenital and cyclic neutropenia occur more frequently in whites compared with individuals of African descent.2 Acute neutropenia is often well tolerated and normalizes rapidly. Neutropenia is often a secondary finding in a patient with far more significant underlying hematologic disorders. Such a patient may be at risk for infectious complications and will likely require thorough investigation. Acute neutropenia evolves over a few days and occurs when neutrophil use is rapid and production is impaired. Chronic neutropenia may last 3 months or longer and arises from reduced production, increased destruction, or excessive splenic sequestration of neutrophils. Neutropenia may be classified by whether it arises secondarily to causes extrinsic to BM myeloid cells, which is common; as an acquired disorder of myeloid progenitor cells, which is less frequent; or as an intrinsic defect affecting the proliferation and maturation of myeloid progenitor cells, which is rare.5
Neutropenia-related infections
Disorders of neutrophil production and release within the BM carry a much higher risk of bacterial and fungal infections than peripheral neutropenia associated with normal BM morphology. The risk for infection in the disorders of neutrophil production and release is greatly increased, with counts of 500-200/μL, and is very severe below 200/μL. The most frequent sites of infection are the skin, mucosa of the oral cavity, and lungs. Disorders of the oral cavity are almost always present by 2 years of age in patients with profound neutropenia associated with myeloid cell production, and are characterized by erosive, hemorrhagic, and painful gingivitis associated with oral ulcers of the tongue and buccal mucosa.3 Occasionally, diffuse gastrointestinal lesions are present, which lead to abdominal pain and diarrhea. These lesions may also be related to bacterial overgrowth in the intestines. Bacterial infections generally involve Staphylococcus aureus, Staphylococcus epidermidis, streptococci, enterococci, Pseudomonas aeruginosa, and other Gram-negative bacilli. Fungal infections usually arise from Candida or Aspergillus species.3 The symptoms of infections may be atypical in patients with neutropenia because there is less local inflammation. Pus and fluctuance may be absent.
The presence of severe neutropenia highlights the critical role of the neutrophil, with its broad array of defense mechanisms that enable it to contain and kill microorganisms. In particular, the neutrophil releases several antibacterial peptides that modulate monocyte chemotaxis and can by themselves can kill bacteria and fungi.6 The NADPH oxidase of neutrophils further serves to kill bacteria and to induce the formation of neutrophil extracellular traps that can contain the spread of bacteria after the intact neutrophils have ceased to function.7 It is not surprising that a lack of circulating neutrophils causes a profound state of immunodeficiency.
Evaluation of neutropenia
Neutropenia is a relatively frequent finding and is often well tolerated and normalizes rapidly, in which case specialized investigations are not necessary. It is sometimes a secondary finding in a patient with far more significant disorders, who may be at risk for infectious complications. More rarely, neutropenia that persists and emerges as the primary cause of symptoms should be investigated thoroughly. The patient's history and physical examination may reveal a particular etiology, such as viral infection, BM malignancy, an iatrogenic cause, immune deficiency, metabolic disorder, autoimmune disorder, or congenital etiology, which warrants further specific investigations (Table 1).4 A more extensive list of rare pediatric etiologies is found in Fioredda et al.4
In a nonurgent setting, the intermittent nature of neutropenia should be established by observing the patient for 6 weeks, obtaining complete counts and differentials 3 times a week and correlating the counts with a patient diary documenting the number of infections, fevers, and any changes in oral health (eg, ulceration and gingivitis).
BM examination is often necessary to exclude malignant pathology in the BM, to determine BM cellularity, and to assess myeloid maturation. Maturation arrest at the promyelocyte stage often occurs, along with BM hypereosinophilia and monocytosis in severe congenital neutropenia (SCN). The BM morphology often does not suggest a particular etiology, with the exception of a few clinical disorders such as Chediak-Higashi syndrome, which is characterized by large cytoplasmic granules; WHIM syndrome (warts, hypogammaglobulinemia, infection, myelokathexis), which is characterized by myelokathexis; or Pearson syndrome, which is characterized by vacuolization of myeloid and erythroid precursors.3 Other investigations helpful in establishing a diagnosis include determination of antineutrophil Abs, Ig assays, lymphocyte immunophenotyping, and reduced levels of the pancreatic enzyme markers isoamylase and trypsinogen (Table 1).8
Humoral immune neutropenia
Neutrophil Ags to which humoral Abs are directed occur in a variety of clinical conditions leading to neutropenia. These include neonatal alloimmune neutropenia, transfusion-related acute lung injury, refractoriness to granulocyte transfusions, febrile transfusion reactions, immune neutropenia after BM transplantation, autoimmune neutropenia, and drug-induced immune neutropenia (Table 2).9,10 The identification of neutrophil Abs is more useful in the pediatric age group in terms of aiding in diagnosis and management. In contrast, the evaluation of systemic autoimmune disease does not require identification of antineutrophil Abs. Tests for antineutrophil Abs parallel RBC serology, with the major exception that the granulocytes in most cases must be fresh. The most widely performed assays for the detection of neutrophil-specific Abs include the granulocyte agglutination test (GAT) and the granulocyte immunofluorescence test (GIFT). The combination of GAT and GIFT appears to be the best means for granulocyte Ab detection. To ensure reliable results, fresh neutrophils from a panel of healthy donors with known phenotypes need to be isolated that cover all or most of the Ag repertoire. Neutrophil Ab testing relies primarily on the reactivity of sera from patients against the neutrophil panel. Both HLA Abs and high levels of immune complexes can lead to false-positive results in both the GAT and GIFT. Both the GAT and GIFT should be performed to confirm the presence of neutrophil-specific Abs. Often, neutrophil Abs are present at low titer and/or bind to the neutrophil-specific Ags with low avidity and may escape detection when only a single attempt is tried. The specificity of neutrophil-specific Abs can be confirmed by an Ag-specific assay such as the mAb-specific immobilization of granulocyte Ag (MAIGA) assay. In contrast to GAT and GIFT, which use intact cells for Ab detection, the MAIGA assay determines only Ab binding to selected glycoproteins present on the granulocyte membrane. The MAIGA assay is rarely used in commercial laboratories.
Granulocyte Ags are called HNA (for human neutrophil alloantigens) to indicate their expression on neutrophils. The glycoprotein location of the Ag is coded by a number; for example, Fc  receptor IIIb is HNA-1. Different polymorphisms of the same glycoprotein are designated alphabetically in sequential order of detection. The human neutrophil alloantigens, their frequencies, and clinical significances are described in Table 2.9,10 The HNA-1 frequencies vary widely among different populations. Among Africans, African Americans, and whites, HNA-1b occurs more frequently than HNA-1a, whereas in Asians (Chinese, Japanese, and Koreans), HNA-1b is found less frequently. The HNA system includes alloantigens for which the main clinical relevance relies on the observation that they are only present on neutrophils and are not expressed in other tissues. Therefore, the highly polymorphic HLAs and the ABO Ags are not part of the HNA systems. In contrast to HLA class-one Ags, the ABO Ags are not expressed on neutrophils. Currently, the HNA system includes 7 Ags that are assigned to 5 Ag groups.
receptor IIIb is HNA-1. Different polymorphisms of the same glycoprotein are designated alphabetically in sequential order of detection. The human neutrophil alloantigens, their frequencies, and clinical significances are described in Table 2.9,10 The HNA-1 frequencies vary widely among different populations. Among Africans, African Americans, and whites, HNA-1b occurs more frequently than HNA-1a, whereas in Asians (Chinese, Japanese, and Koreans), HNA-1b is found less frequently. The HNA system includes alloantigens for which the main clinical relevance relies on the observation that they are only present on neutrophils and are not expressed in other tissues. Therefore, the highly polymorphic HLAs and the ABO Ags are not part of the HNA systems. In contrast to HLA class-one Ags, the ABO Ags are not expressed on neutrophils. Currently, the HNA system includes 7 Ags that are assigned to 5 Ag groups.
Cell-mediated neutropenia
Chronic idiopathic neutropenia in adults is usually benign and uncomplicated and is characterized by the absence of antineutrophil Abs.11 A less common form affects mostly middle-aged women. It is characterized by mild neutropenia for more than 3 months with an ANC < 1500/μL for white subjects. There is an absence of clinical, serologic, or ultrasound evidence of any underlying disease associated with neutropenia. Often, mild anemia and/or thrombocytopenia may accompany the neutropenia. BM cellularity is usually normal; however, mild hypoplasia of the myeloid series with a shift to the left may be present. In the BM, there is an increased proportion of T lymphocytes in a predominantly interstitial pattern, less frequently in a nodular pattern. The presence of activated nonclonal T lymphocytes expressing high levels of HLA-DR, CD25, CD38, CD69, and Fas has been observed in both the blood and BM. The T lymphocytes are the source of IFN-γ and Fas-ligand, which play a myelosuppressive role in coculture experiments in vitro. In contrast, the etiology of more common forms of chronic idiopathic neutropenia in both pediatric and adult patients lacking T lymphocytes remains unknown.2
Large granular lymphocyte (LGL) leukemia is a clonal disease representing a spectrum of biologic distinct lymphoproliferative diseases originating either from mature CD3+ T cells or CD3− natural killer cells.12 LGL is primarily a disease of adults. Both CD3+ and CD3− LGLs function as cytotoxic lymphocytes. LGL leukemia is diagnosed in a clinical spectrum of cytopenias, lymphocytosis, splenomegaly, and autoimmune conditions such as rheumatoid arthritis. Patients with Felty syndrome and T-cell LGL with rheumatoid arthritis share a similar frequency of the HLA-DR4 allele (80%-90%), whereas those without rheumatoid arthritis do not and are similar to racially matched controls (33%). Patients with LGL leukemia have more than 500/μL LGL cells expressing CD3+, CD16+, CD28−, and CD57+ in their peripheral blood. The main criteria for diagnosis of LGL leukemia is the detection of a clonal TCR rearrangement with a typical phenotype of TCR-alpha, beta. Neutropenia is the most common finding in LGL leukemia and occurs in 70%-80% of patients. Neutropenia in LGL leukemia results from impaired production in the BM through cell-mediated mechanisms and increased neutrophil destruction mediated by humoral mechanisms. Mild to moderate splenomegaly occurs in 20%-60% of LGL leukemia patients, but the degree of splenomegaly is not correlated with the hematologic abnormalities, including neutropenia. Several lines of evidence suggest that clonal expansion of LGL leukemia may be Ag driven. Members of the HTLV genus of retroviruses such as Human T-Lymphotrophic Virus type 1 (HTLV-1) and bovine leukemia virus (BLV) are often detected serologically in patients with LGL. After Western blot testing against HTLV-1 viral lysate, LGL leukemia cells show a similar cross-reactivity pattern upon testing with HTLV-1 or the BLV + serum, which further implicates a viral etiology.
Drug-induced neutropenia
Drug-induced neutropenia is an adverse event resulting in an ANC below 500/μL. It is associated with a high rate of infectious complications and has a mortality rate ranging from 2.5%-10%.13 The highest mortality rate is observed in older patients and in those experiencing renal failure, bacteremia, or shock at diagnosis. The incidence increases with age, because only 10% of cases are reported in children and young adults and half of these episodes occur in subjects over 60 years of age, which likely reflects higher use of multiple medications in elderly people. Almost all classes of drugs have been implicated, but the risk appears to be very small for the majority of these compounds. The most common drugs associated with severe neutropenia are antithyroid medications, ticlopidine, clozapine, sulfasalazine, trimethoprim-sulfamethoxazole, and dipyrone.14 The mAb anti-CD20 (rituximab) also causes late-onset neutropenia.15 If a potential causative agent is identifiable, drug-induced neutropenia is sometimes reversible at withdrawal of the suspected drug, thus enabling diagnosis and treatment.
The pathogenesis of drug-induced neutropenia is heterogeneous and is not completely understood. In some cases, neutropenia occurs after prolonged exposure to drugs, resulting in decreased myeloid production from a hypoplastic BM.13 Other cases occur after repeated but intermittent exposure to offending agents. This suggests an immune mechanism and, in some cases, antineutrophil Abs are found arising from both autoantibodies and drug-dependent Abs.
Clozapine-induced neutropenia occurs in approximately 1% of patients, particularly in the first 3 months of treatment.16 The occurrence of neutropenia appears to be associated with distinct histocompatibility Ags. Clozapine-induced neutropenia is thought to arise from a depletion of ATP and reduced glutathione, which renders the neutrophil susceptible to oxidant-induced apoptosis.
Disorders of production secondary to genetic etiologies
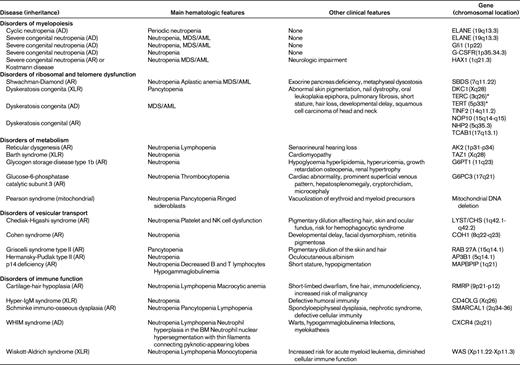
Genotype is the most important factor for distinguishing one form of congenital neutropenia from another, but it is usually not available during the initial evaluation. The phenotype represents a continuum that may develop fully with age. Overlap between clinical manifestations and some important organ involvement may not be observed at the initial evaluation. For example, with Shwachman-Diamond syndrome, neutropenia may be the initial observation, but a patient may develop other cytopenias and aplastic anemia over time. Table 3 shows the different subgroups with their known genes. Table 4 indicates the normal function of genes found in chronic neutropenia disorders.
Cyclic neutropenia
Cyclic neutropenia is a rare, autosomal-dominant disorder arising from mutations in the gene for neutrophil elastase (ELANE or ELA-2) observed in 80% of affected subjects and occurs with a frequency of 0.6/million persons.3,17 These patients commonly have regular oscillations of peripheral blood neutrophil counts with periods of severe neutropenia lasting for 4-6 days and occurring every 21 days. During the periods of profound neutropenia, the patients are predisposed to developing painful mouth ulcers, fever, and bacterial infections. Children are particularly at risk for developing severe consequences of profound neutropenia, including gangrene, bacteremia, and septic shock.18 Most mutations in ELANE found in cyclic neutropenia are usually confined to exons 4 and 5, but there can be overlap of gene mutations in congenital neutropenia patients. It is imperative to establish the diagnosis of cyclic neutropenia by serial differential white counts at least 3 times per week for a minimum of 6 weeks to observe at least 2 neutrophil nadirs. Such an approach will help to differentiate the disorder from SCN that may at times share the same ELANE mutation. Cyclic neutropenia, unlike the autosomal-dominant form of congenital neutropenia associated with mutations of ELANE, is not associated with an increased risk for leukemia or myelodysplasia. This information is obviously important to share with the affected patient and family. Figure 1 illustrates the pattern in a patient thought to have cyclic neutropenia and contrasted with an individual with classic cyclic neutropenia. As seen in Figure 1, the patient with cyclic neutropenia has a reciprocal rise in the monocyte count at the time of the nadir of the neutropenia. In contrast, the monocyte count is variably elevated in congenital neutropenia and not in a reciprocal fashion with the nadir of the neutrophil count.

Pattern in a patient thought to have cyclic neutropenia compared with an individual with classic cyclic neutropenia. (Left) ANC and absolute monocyte count in a patient with congenital neutropenia over time. (Right) ANC and absolute monocyte count in a patient with cyclic neutropenia over time.
Pattern in a patient thought to have cyclic neutropenia compared with an individual with classic cyclic neutropenia. (Left) ANC and absolute monocyte count in a patient with congenital neutropenia over time. (Right) ANC and absolute monocyte count in a patient with cyclic neutropenia over time.
SCN and Kostmann disease
SCN was initially described by Kostmann as an autosomal-recessive disorder in an isolated population in Sweden.19 Other forms of SCN have been identified with sporadic occurrence or with autosomal-recessive or autosomal-dominant inheritance. We suggest that the term SCN should refer to the entire disorder and that Kostmann disease refer to the autosomal-recessive subtype.
SCN is characterized by ANCs consistently below 200/μL with recurrent severe infections often developing in the first months of life.18 BM examination characteristically shows a myeloid “maturation arrest” at the promyelocyte-myelocyte stage of development. The apparent maturation arrest helps to differentiate SCN from idiopathic and immune neutropenia. Many patients left untreated suffer from chronic gingivitis, oral ulcers, skin abscesses, recurrent pneumonia, or septicemia.
After the discovery of mutations in the ELANE gene in patients with cyclic neutropenia,17 ELANE mutations were observed in patients with SCN and they were inherited in an autosomal-dominant manner.20,21 More than 50 defined mutations have been recognized to be associated with cyclic neutropenia or SCN.22 SCN has a rate of 2/million persons.3 ELANE mutations are found in approximately 40%-60% of patients with congenital neutropenia. Compared with other forms of congenital neutropenia, neutropenia due to ELANE mutation is associated with the most severe infectious complications. The same mutations can be responsible for both types of severe neutropenia, cyclic and congenital neutropenia. These 2 subtypes are considered as part of a continuum of the same disease. This has been well documented in cases of children conceived by artificial insemination or in vitro fertilization from the same sperm donor, who were found to have congenital neutropenia or cyclic neutropenia.23
ELANE mutations lead to severe neutropenia via a stress response in the endoplasmic reticulum (ER), which provokes activation of the unfolded protein response (UPR).24,25 The UPR has evolved to protect cells from the damaging effects of improperly folded proteins. Myeloid cells destined for secretory vesicles are directed to the ER, where protein folding takes place. In the case of abnormal protein folding, ER stress occurs, leading to activation of 3 ER-localized protein sensors: inositol-requiring 1-α (IRE1-α), PRK-like ER kinase (PERK), and activating transcription factor-6 (ATF6). When proteins are misfolded, the sensors are activated and trigger a complex series of events designed to maintain the homeostasis of the ER and to promote proper protein folding, maturation, secretion, and ER-associated protein degradation. Should the rescue mechanisms fail, the UPR leads to apoptosis to protect cells from dysfunction. Both myeloid cell lines and primary myeloid human cells from SCN patients with ELANE mutations show increased biochemical evidence of ER stress and activation of UPR sensors, which in part explains the increased apoptosis of myeloid progenitor cells. The heightened apoptosis of myeloid precursors is reflected in the characteristic BM morphology. Other very rare causes of autosomal-dominant SCN arise from Gfi1 mutations mediating transcriptional repression of myeloid genes, production of T lymphocytes, and an inherited mutation of the extracellular domain of the G-CSF receptor gene.26,27
Mutations in most autosomal-recessive SCN kindreds include several described by Kostmann affecting the HAX1 gene, which encodes a mitochondrial protein.28 Both hematopoietic and nonhematopoietic cells in HAX1 deficiency are susceptible to induced dissipation of the inner mitochondrial membrane potential, leading to accelerated cell apoptosis. Descendants of families originally described by Kostmann have been noted to suffer from cognitive disorders. Other SCN patients with HAX1 mutations exhibit mild developmental delay to severe epilepsy. These observations led to the identification of 2 human HAX1-spliced variants consisting of isoform A and isoform B. Isoform B is characterized by a splice event removing part of exon 2 and is preferentially expressed in normal cells. Mutations affecting only isoform A lead to a phenotype restricted to congenital neutropenia, whereas mutations affecting both isoform A and isoform B are associated with the phenotype of congenital neutropenia and various degrees of neurologic impairment.22
Neutropenia occurring with complex phenotypes have been clarified by the identification of underlying genetic defects. We have suggested29 the resultant classification of the syndromes be categorized into disorders of ribosomal dysfunction (Shwachman-Diamond syndrome and dyskeratosis congenita)30,31 ; of metabolism (reticular dysgenesis, Barth syndrome, glycogen storage disease type 1b, and glucose-6-phosphatase catalytic subunit 3 syndrome)32–34 ; of vesicular transport (Chediak-Higashi syndrome, Cohen syndrome, Griscelli syndrome type II, Hermansky-Pudlak syndrome type II, and p14 deficiency);35–39 and disorders of immune function (cartilage-hair hypoplasia, hyper-IgM syndrome, Schimke immuno-osseous dysplasia, WHIM syndrome, and Wiskott-Aldrich syndrome).3,21,40,41 The spectrum of congenital disorders of neutropenia and their genetic diagnosis are described in Table 3.
Leukemia risk
The introduction of growth factors such as G-CSF in the late 1980s vastly improved the management of chronic neutropenia. The need for long-term administration of G-CSF was clear but the question of safety was raised. This led to the formation of the Severe Chronic Neutropenia International Registry (SCNIR). Data from SCNIR confirmed an increase of leukemia, especially in those patients with SCN arising from mutations in ELANE and HAX1. The cumulative incidence of leukemia among patients with SCN has ranged from 10%-20% and equally affected those with and without ELANE mutations after 15 years of treatment.42 The progression to myelodysplasia and/or acute myelogenous leukemia is suggested by altered complete blood counts (CBC) and confirmed by BM examination and BM cytogenetics indicating an increased number of blasts or at times acquired monosomy 7 or trisomy 21 genotype. At least quarterly CBCs and annual BM aspirations with cytogenetics should be performed to adequately follow SCN patients. The requirement for doses of G-CSF exceeding 8mcg/kg is an indicator of risk, likely indicating a more severe disease, although a contributing contribution of G-CSF to the risk in not excluded. Hence changes in the response to G-CSF or other alterations in the blood counts should suggest a need for a BM. Leukemic transformation has also been observed in patients with Wiskott-Aldrich Syndrome, Shwachman-Diamond Syndrome, and glycogen storage disease 1b.3 The leukemic transformation in SCN appears to follow sequential gain of mutations leading to the leukemia phenotype.43
Summary
In conclusion, a better understanding of the molecular basis of various congenital neutropenia disorders and the normal function of the involved genes has provided insights into the biology of the myeloid cell (Table 4). The genetic mutations have involved membrane structures, secretory vesicles, mitochondrial metabolism, ribosome biogenesis, transcriptional regulation, and cytoskeletal dynamics. Therefore, the unraveling of these genetic disorders has contributed to our improved diagnostic acumen regarding the etiology of the chronic neutropenia disorders and the risk for myelodysplasia/acute myeloid leukemia.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from the National Institutes of Health, has been affiliated with the speakers' bureau for Alexion, holds patents with or receives royalties from Up to Date, and has equity ownership in Amgen. Off-label drug use: None disclosed.
Correspondence
Laurence A. Boxer, Department of Pediatrics and Communicable Diseases, University of Michigan Health System; D3251 MPB, 1500 E Medical Center Dr, Ann Arbor, MI 48109-5718; Phone: 734-764-7126; Fax: 734-615-0464; e-mail: laboxer@umich.edu.