Abstract
Allogeneic HSCT controls sickle cell disease (SCD)–related organ damage and is currently the only curative therapy available. Over the last 2 decades, HSCT has been limited largely to myeloablative matched sibling donor (MSD) procedures that are feasible only in a minority of patients. As the natural history of the disease has evolved, it is clear that subsets of patients with severe disease are at risk for sudden death, devastating CNS and pulmonary complications, and debilitating vasoocclusive crises. For these patients, the benefits of transplantation can outweigh the risks if HSCT can be safely and successfully performed with low early and late toxicities. This review describes advances and ongoing investigation of HSCT for SCD from the perspectives of recipient age and presentation, donor stem cell source, intensity of conditioning, family and medical perspectives, and other variables that influence outcome. Ultimately, HSCT should be viewed as a viable treatment option for SCD on par with other therapies for select patients who can benefit from the procedure.
Introduction
Sickle hemoglobin (HbS) results from an amino acid substitution of glutamine to valine at the sixth position in the beta-globin chain as a result of a single nucleotide mutation. Homozygosity (HbSS) results in sickle cell disease (SCD). HbS can occur in combination with other hemoglobin mutations, resulting in disease variants such as HbSC disease or HbS-β thalassemia. Although HbSS and Sβ0 thalassemia are traditionally implicated in the most severe forms of the disease, other variants are not immune from clinical complications that can be life-threatening or debilitating. Polymerization of HbS in deoxygenated states distorts red cells into sickled shapes, resulting in hemolysis and anemia. The widely variable severity of symptoms and target organ damage even within similar phenotypes depends on the extent of vascular inflammation/injury, cellular/chemokine changes, and the induction of a hypercoagulable state that is blamed on inflammatory pathways spurred by nitric oxide depletion, C-reactive protein, inflammatory cytokines such as IL-6, and adhesion molecules.1,2 However, the level of contribution of individual pathways is unclear, making targeted pharmacologic intervention complicated.3
Although not curative, there have been significant advances in supportive care for SCD in the last decade. The most influential have been symptom control, primarily by hydroxyurea, and increased sophistication in red cell transfusion therapy, such as extended phenotype matching to reduce alloimmunization and erythrocytapheresis to reduce iron overload. Hydroxyurea has provided significant benefit by reducing pain episodes, acute chest syndrome hospitalizations and transfusions, and mortality rates both in adults and children, and has minimal genotoxicity or carcinogenicity despite long-term use.4
Only successful HSCT or gene therapy with transduced autologous hematopoietic stem cells can establish complete or partial normal erythropoiesis that results in a cure. Because gene therapy is still in its early development, HSCT is the only established curative treatment modality. HSCT can establish donor-derived erythropoiesis and, more importantly, can stabilize or restore function in affected organs and prevent further morbidity. Although the benefits of HSCT are well established, complications associated with the procedure require continued investigation to target goals such as determining the population best benefited by the intervention, improving engraftment, reducing post-HSCT toxicities and GVHD, and improving availability (donors). These efforts should proceed in parallel with other intervention and supportive care trials in afflicted individuals.
Assessing indications and eligibility for HSCT
As in all nonmalignant disorders, for best outcomes, HSCT for SCD should be performed when a recipient is at a good functional baseline in anticipation of the inevitable organ damage that is driven by the duration of pathology—and thus age. However, clinical variability in time of onset and the eventual degree of morbidity and severity of organ involvement pose a unique problem in defining both eligibility and ideal timing for HSCT. Attempts at defining early predictors of future severity using markers such as leukocytosis or clinical symptoms have proved largely inaccurate.5
CNS involvement defined by overt stroke or high transcranial Doppler velocities is a definitive indicator of continued risk for recurrent CNS events.6 The need for lifelong erythrocytapheresis to attempt to offset progression suggests that these patients are best served by transplantation. The incidence of overt stroke in SCD is 9% by 14 years of age.7 Another 18% develop MRI changes consistent with silent cerebral infarcts by this age: a 27% rate of neurologic complications before adolescence.8 Up to 20% of children with previous strokes and cerebral vasculopathy can experience second strokes within 5 years despite adequate transfusion therapy.9 New onset cerebral infarction (overt or silent) was noted in as many as 45% of chronic blood transfusion therapy recipients, which underscores the need for more effective interventions.10 The cumulative risk of all cerebral disease by age 14 is a high 50%, a risk mitigated by HSCT.11,12
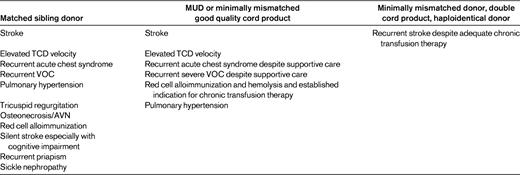
Cardiopulmonary events, primarily acute chest syndrome and pulmonary hypertension, account for > 50% mortality in young adults with SCD.13,14 Stability of pulmonary function is achieved with HSCT. Tricuspid regurgitation measured as a precursor to pulmonary hypertension was noted in more than 20% of children at a mean age of 6.2 years.15 The debilitation of recurrent vaso-occlusive crises is harder to define precisely for a transplantation threshold, but they significantly impair quality of life.16 Other severe complications include sickle nephropathy, avascular necrosis, silent stroke with cognitive impairment, priapism, and red cell alloimmunization. HSCT needs to be weighed in the context of the risk-benefit ratio for each of these indications. Similarly, stringency regarding indications is advisable in the absence of HLA-matched sibling donors (MSDs) given the difference in expected outcomes—especially from the perspective of morbidity/mortality and GVHD. HSCT indications should remain a fluid paradigm revisited intermittently based on changing transplantation protocols and outcomes (Table 1). With stringent severity criteria (CNS and pulmonary), approximately 16% of patients would qualify for transplantation; approximately 38% would fit extended criteria.17 A case could be made for consideration of extended criteria for related donor HSCT, reserving unrelated donor (URD) transplantations only for those patients with severe disease based on stringent criteria. Donor availability and transplantation indications (Table 1) need to be further balanced with recipient age because increased morbidity and mortality is expected with young adult patients compared with HSCT in children with SCD. This is attributable to established organ dysfunction and comorbidities expected with age. Because the validity of expanding HSCT indications are defined by outcomes to define risk-benefit ratios, expansion of transplantation options and donor options from the sibling to the URD setting should only be undertaken in a trial setting.
Scope of the disease in relation to transplantation and timing
There are more than 70 000 individuals with SCD living in the United States (1000 SCD births per year). Approximately 94% of children with HbSS or HbSβ0 thalassemia will reach 18 years of age, but stroke-free survival decreases to 88.5%.18 The average life expectancy for male and female patients with SCD has been determined to be 42 and 48 years, respectively.19 HSCT is currently an underutilized intervention given the prevalence of the disease. Only ∼ 450 transplantation procedures are reported in the Center for International Blood and Marrow Transplant Research database, of which 85% are from MSDs and more than 80% are in children less than 16 years of age. Disease and age-related comorbidities result in higher morbidity and mortality rates in older patients undergoing HSCT.20,21 Management of disease-related organ damage exacerbated by HSCT-related conditioning and other events is complex and SCD transplantations have unique complications that need to be managed to prevent excess morbidity and mortality; these include hypertension, stroke, seizures, and renal dysfunction. Because transplantation is generally undertaken after an established complication predicts severe disease, almost all recipients are higher risk. Myeloablative transplantation outcomes are good in the young (< 16 years) likely due to the absence of comorbid conditions and organ decompensation.22–24 This age bar could potentially be pushed higher into the second decade with a reduced-intensity treatment (RIT) conditioning approach that could be better tolerated in the presence of organ toxicity. Trials of RIT HSCT that include older patients with SCD using matched related, unrelated, and haploidentical donors are under way and in early stages of enrollment at the National Institutes of Health, Blood and Marrow Transplant-Related Clinical Trials Network (BMT CTN) Sickle Cell Unrelated Transplant (SCURT), the NIH, and at the Johns Hopkins School of Medicine.
Acceptability of transplantation in the pediatric age group is a parent-dependent decision that can be a difficult even with adequate information and education. When SCD parents were polled by questionnaire, 37% of parents were willing to accept HSCT as a treatment modality given a 15% mortality risk, but acceptance dropped to 13% if the mortality risk was 15% and GVHD risk was 15%.25 The acceptability of transplantation is higher in adult SCD patients, presumably because of experience with complications of the disease. In 2 adult surveys, 63% of adult SCD patients were willing to accept a mortality risk of > 10% and 60% were willing to accept a RIT transplantation trial, but only 20% would accept chronic GVHD and only 50% would accept infertility.26,27
Donor sources determine ability to offer HSCT
Fewer than 14% of SCD patients have MSDs. Patients with sibling donors can consider transplantation early in the disease process due to good outcomes. This option may be even more attractive if the patient is returning to an area with poor access to supportive care. In the absence of a MSD, a matched URD (MUD) HSCT must be considered in the presence of severe disease criteria. This procedure used to be rarely considered by the hematology community due to toxicity and mortality risks, but is now more acceptable with RIT regimens that are better tolerated.28 The paucity of minority donors in the registry often limits finding a suitable MUD in this population.29 The probability of finding a matched marrow or cord product for SCD patients depends on the level of mismatching that is considered acceptable. Restricting donor searches to 8 of 8 high-resolution HLA matching for BM and 5-6 of 6 intermediate-resolution (class I) and high-resolution (class II) for cords with an adequate cell dose (> 5 × 105 CD34/kg) limits availability to approximately 30%-40%.30–32 Upon identification, donor withdrawal has also been a problem. Donor solicitation and retention are critical for the success of HSCT in minority populations irrespective of disease indication.
As treatment strategies continue to improve, single allele–mismatched marrow transplantations and matched or minimally mismatched single and double cord transplantations with optimized cell dose requirements may be considered acceptable, especially if HSCT methods provide more safety. As an example, strategies such as immunoablative regimens with alemtuzumab that can partially deplete immunoreactive graft T cells or posttransplantation cyclophosphamide as GVHD prophylaxis are under evaluation.33,34
Mobilized peripheral blood products have a higher risk of chronic GVHD and mortality, especially in children. For this reason, pediatric transplantation protocols that use unmanipulated stem cell products have preferred to use BM, especially in nonmalignant disorders whenever feasible. Although chronic GVHD rates are no different, adult patients have had higher mortality after related donor peripheral blood stem cell transplantations for severe aplastic anemia, and may also benefit from marrow as the stem cell source.35 Haploidentical transplantations (preferably from the mother due to the possibility of tolerance to maternal antigens) are under investigation with CD34 selected products and early success has been reported.33 With trials continuing to investigate and define outcomes in a controlled manner, a future reality will be the ability to offer HSCT to eligible patients based on a selection algorithm (Figure 1).

Algorithm for consideration to expand donor selection on experimental HSCT trials for SCD. The efficacy of MSD transplantations and the paucity of sibling donors requires continued careful evaluation in trial settings of alternative donor transplantations; options are listed in this algorithm.
Algorithm for consideration to expand donor selection on experimental HSCT trials for SCD. The efficacy of MSD transplantations and the paucity of sibling donors requires continued careful evaluation in trial settings of alternative donor transplantations; options are listed in this algorithm.
Myeloablative transplantations: one size does not fit all
Successful myeloablative transplantation trials in children from HLA MSDs using busulfan-based preparative regimens (busulfan, cyclophosphamide ± anti-thymocyte globulin, or anti-lymphocyte globulin ± total lymphoid irradiation) have been reported in the past 10-15 years.22,24,36 Combined outcomes are listed in Table 1. Overall survival is > 90% and event-free survival > 80%. Although fewer in number, myeloablative sibling donor cord blood transplantations have also produced excellent results in children (Table 1).37,38 Most importantly, as disease parameters were followed after engraftment, the majority (> 90%) were relieved of pain, had no further strokes or acute chest syndrome, had stabilized pulmonary function, and had stable neurologic and cognitive evaluations.12 However, on cerebral imaging, new MRA/MRI changes were noted after HSCT in patients with previous MRI abnormalities (lacunae/infarcts); stroke has also been reported after HSCT.39,40 If underlying vasculopathy is to blame, additional stressors such as toxicities from the conditioning, cytokine storm and engraftment syndrome, hemodynamic changes of HSCT, and GVHD-related complications could be a negative influence. The age and eligibility for transplantation, conditioning strategy, management of SCD patients after HSCT, and tailoring transplantation strategies based on donor availability and recipient status are the focus of newer transplantation trials and worthy of investigation.
Previous trials have been instrumental in proving the benefit of transplantation in SCD. Deterrents to HSCT include the risk of immediate mortality, graft rejection, infections, GVHD, and later sterility, ovarian failure, and poor spermatogenesis. Supportive care strategies such as hydroxyurea and erythrocytapheresis/iron chelation therapy have been perfected to support reasonable longevity into middle adulthood, but the patient's course will be difficult due to the medical and psychosocial complications of chronic disorders such as iron overload, infection risk, alloimmunization, narcotic dependence, poor compliance, and poor quality of life. The cost and availability of medical resources for lifelong medical care has to be considered. Hospital admissions cost $1000/day and outpatient visits for transfusions cost $350 each (based on 2009 data). Further, disease progression, organ involvement, and sudden death can occur despite these measures. Before embarking on HSCT, it is essential that both conservative therapy options and HSCT pros and cons are discussed in detail and in a balanced fashion by a combined hematologist-transplantation team for informed decision making. Like supportive care measures, transplantation efforts targeting newer interventions are directed at improving the safety and efficacy of HSCT to minimize areas of risk and make a curative intervention available to the majority of patients.
Mixed chimerism is acceptable in nonmalignant disorders
Stable mixed donor-host chimerism is adequate to establish a cure. Donor chimerism levels can vary widely (11%-74%) and yet still support normal HbA levels after HSCT for SCD.22 The level of circulating HbS is determined more by donor genotype than level of chimerism. Recipients of HSCT from donors with sickle trait (chimerism levels of 25% and 60%) had higher HbS (36% and 37%) than those receiving transplantations from normal donors. When donor chimerism has been 67%, 74%, and 11% in recipients of normal donor transplantations, HbS levels of 0%, 0%, and 7%, respectively, were noted. Donor chimerism as low as 11% resulted in only 7% circulating HbS and sustained clinical improvement. This redefines graft rejection and rejects the conventional definition of the term.22 Late graft rejection beyond the first year is always a concern but has not proven to be an issue in myeloablative SCD transplantations. In thalassemia transplantations, mixed chimerism was noted in 74% and 34% of recipients at 1 and 6 months, respectively; the incidence of secondary (late) graft rejection at 1 year after transplantation was only 8%.41 Murine models of SCD suggest that normal erythrocytes have a survival advantage that may be based both on normal red cell longevity and effective erythropoiesis.
RIT and immunoablative HSCT
With the acceptance of stable mixed chimerism as curative, it is easier to explore the possibility of RIT conditioning in an attempt to limit acute and long-term organ toxicities of myeloablation and offer the HSCT option to older patients who would not have tolerated conventional transplantations. RIT includes multiple strategies such as using conventional agents at lower doses or resorting to targeted immune ablation. Because outcomes vary based on strategy, all RIT should not be considered equal. RIT procedures are still under evaluation and outcome data are early.
There are some lessons learned previously. Early trials in SCD patients have demonstrated a “threshold balance” between intensity of conditioning, stem cell source, and posttransplantation immune suppression—a work in progress. In addition to stem cell source and quality, the balance between intensity of conditioning and engraftment is influenced by host factors such as transfusion history, alloimmunization to red cell and HLA antigens, and blood group incompatibility. Outcomes are summarized briefly in Table 2. In general, cord transplantations require higher-intensity conditioning than BM transplantations. Nonablative regimens based on low-dose radiation (200-400 cGy) and fludarabine have not supported engraftment.44,45
Regimens targeting immune ablation with combinations of alemtuzumab, fludarabine, melphalan, or low-dose busulfan, total lymphoid irradiation, and Cytoxan have been successful in ameliorating disease.28,46 Our preliminary experience with 14 patients treated in a multicenter trial with a combination of alemtuzumab, fludarabine, and melphalan (using MRD or MUD marrow) had an overall survival of 95% and event-free survival of 79% in patients who were at least 6 months posttransplantation (unpublished data). High-dose CD34-selected cells from HLA-matched siblings has supported mixed chimerism after alemtuzumab and low-dose total body irradiation conditioning and sirolimus posttransplantation.47 The sirolimus was targeted at increasing T-regulatory cells to induce long-term tolerance. Continued immunosuppression was able to maintain mixed lymphoid chimerism status in the sibling donor setting and an investigation of delayed sirolimus wean is under way.
URDs, cord blood, and haploidentical transplantations
Although the benefits of MSD transplantation are well described, URD transplantation has been rarely used in SCD. However, URD HSCT should remain a consideration, especially in those with severe disease, extrapolating from URD transplantations described for thalassemia. Disease-free survival rates of 70%-80% have been described in good-risk thalassemia patients, especially in those with extended HLA matching; as expected, those in poor-risk groups (Pesaro class III) fared less well.48–50
Cord blood, by virtue of allowing more mismatching and targeted increase in minority collections of stem cell product, are attractive as a focus for the development of safer and more successful transplantation designs. The poor outcomes of URD myeloablative cord transplantations (Table 2) suggest that new avenues of investigation are necessary.51,52 URD cords have a high risk of rejection with RIT, and more intensity with RIT before cord transplantation is under evaluation for hemoglobinopathies at our and other centers. Cord cell dose is critical, and a cell dose of > 5 × 105 CD34 cells/kg is recommended in nonmalignant disorders with a high rejection rate. More recently, total CFUs have been identified as even more useful in predicting cord engraftment and may become the standard assessment of the future.53 Emerging data on double cord transplantations and successful engraftment, even in adult patients, open another avenue of circumventing engraftment barriers.54 Success with ex vivo cord cell expansion may be a reality in the near future.
Although almost all SCD patients are likely have a haploidentical donor, HSCT studies in SCD have been rare due to the risk of GVHD, rejection, and poor immune reconstitution. Haploidentical CD34-selected HSCT using pretransplantation fludarabine, Cytoxan, and low-dose total body irradiation with higher-dose Cytoxan after transplantation as GVHD prophylaxis has had early success without evidence of GVHD.33 Continued success with this approach opens yet another transplantation avenue for SCD.
Taking host factors (disease severity) and donor availability into consideration, a donor selection algorithmic process with optimized individual transplantation approaches for each source is a necessary future direction that will allow the best use of an available stem cell source (Figure 1). A single transplantation approach will not fit all. Similarly, the acceptability of adverse outcomes such as treatment-related mortality, graft rejection, and chronic GVHD will need to vary between groups. Figure 2 shows anticipated differences in HSCT outcomes after URD transplantations based on host factors.

Conceptual projection of acceptable transplantation toxicities based on known risks from URD transplantation.
Conceptual projection of acceptable transplantation toxicities based on known risks from URD transplantation.
The BMT CTN is conducting a trial of URD BM RIT transplantation, the SCURT trial, for children less than 20 years of age with severe SCD. This trial accepts only fully (8 of 8 at A, B, C, and DRB1) matched marrow donors to optimize outcomes. Stopping rules are designed to detect unacceptable outcomes of death, GVHD, and graft rejection in a timely fashion. This is an important trial designed to answer questions regarding the benefits of URD HSCT for SCD. Our current strategy in children is to use an RIT regimen (alemtuzumab, fludarabine, and melphalan) for patients with matched marrow donors and to enroll eligible patients in the SCURT trial. This trial was closed to unrelated cord transplantation recipients due to increased graft rejection.
Patients with severe disease with unrelated, well-matched cord/double cord/allele mismatched marrow options can be enrolled in other experimental multicenter trials of intensified RIT conditioning, with requisites for cord size and matching. Haploidentical CD34-selected stem cell transplantation trials are in development or under way at several centers, as are RIT transplantations for young adults. Ultimately, the cooperative goal of the sickle cell transplantation community is to design safe and beneficial transplantation approaches that can cure this devastating disease. These need to be evaluated in a trial setting so that outcomes can be clearly defined.
Whither to SCD transplantations?
SCD transplantations have arrived, but need to expand in applicability in a trial setting. With previous studies, there is increased recognition of transplantation pros and cons based on disease, age, and timing of transplantation, donor source, toxicities of conditioning therapy, etc. To continue to improve upon success, safety, and tolerability, new approaches need to be evaluated, especially for URD HSCT, to reduce GVHD and toxicities and to enhance engraftment. A team approach in which hematologists, health care staff, transplantation physicians, patients, and families work together to offer the most appropriate individualized care to the SCD patient is necessary. Transplantation options need to be accepted as one of the interventions that can be beneficial if indicated. This includes an early assessment for MSDs for which transplantation may be preferable even with “milder” forms of the disease given the successful outcomes. Alternative donor transplantations should be reserved for more severe presentations. The field of SCD transplantations must continue to advance with formal transplantation studies so that the best-available HSCT option may be offered to all patients who can benefit from the intervention.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Shalini Shenoy, MD, Division of Pediatric Hematology/Oncology, Box 8116, Washington University, St Louis Children's Hospital, 1 Children's Place, St Louis, MO 63110; Phone: (314) 454-6018; Fax: (314) 454-2780, e-mail: shenoy@wustl.edu.